










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActas Urológicas Españolas
versión impresa ISSN 0210-4806
Actas Urol Esp vol.30 no.4 abr. 2006
Aplicación clínica de las actuales clasificaciones del cáncer renal
Usefulness of the present renal cell carcinoma classifications
F. Algaba1,2, Y. Arce1, Trias I.3, J.M. Santaularia1, A. Antonio Rosales4
1Sección de Patología, Fundació Puigvert- Barcelona.
2Departamento de Ciencias Morfológicas de la Facultad de Medicina de la Universitat Autónoma de Barcelona (UAB).
3Departamento de Patología, Clínica Plató-Fundació Privada- Barcelona.
4Servicio de Urología, Fundació Puigvert-Barcelona.
Dirección para correspondencia
RESUMEN
De forma ideal la finalidad de las clasificaciones de las neoplasias es el reconocer grupos homogéneos de evolución y pronóstico y si es posible de igual tratamiento. Este es el motivo por el que de forma sistemática se van revisando y puliendo a través de las nuevas metodologías.
En el cáncer renal la discriminación de diferentes entidades ha sido relativamente rápida ya que los nuevos hallazgos genéticos y moleculares han reafirmado los criterios morfológicos de los distintos tipos celulares, y posiblemente se están abriendo nuevas vías terapéuticas.
Con la actual clasificación de la OMS se reconocen subtipos con un excelente pronóstico (Carcinoma renal quístico multilocular, Carcinoma papilar renal de tipo 1, Carcinoma mucinoso tubular y fusocelular), otros muy agresivos (Carcinoma de ductos colectores de Bellini, Carcinoma medular) y que la transformación sarcomatoide, aún en pequeñas áreas, tiene un impacto negativo sobre el pronóstico. Se han reconocido los carcinomas renales asociados a translocaciones cromosómicas (fusión genética TFE3 o TFEB) propios de la infancia y las distintas formas de carcinoma renal familiar.
En cuanto a la clasificación de la UICC, hay una serie de aspectos en controversia (tamaño, invasión venosa, invasión microvascular, invasión del tejido adiposo del seno renal) que también son comentados, ya que quizás en un futuro próximo pueden haber modificaciones del TNM.
Palabras clave: Carcinoma renal. Carcinoma células claras. Carcinoma renal papilar. Carcinoma de células cromófobas. Carcinoma Bellini. Carcinoma renal medular- Carcinoma renal nuevas entidades.
ABSTRACT
The purpose of classifying neoplasias is to recognize groups with similar progress and prognosis and, if possible, receiving the same treatment. This is why those classifications are systematically being submitted to review and improvement through the new technologies.
Differentiation of various entities in renal cancer has been comparatively fast, as the new genetic and molecular discoveries have confirmed the morphologic criteria of the different cell types, thus making it possible to open new therapeutic pathways.
Using the current WHO classification we recognize subtypes with excellent prognosis (Multilocular cystic renal carcinoma, Type I renal papillary carcinoma, Tubular and fusocellular mucinous carcinoma), other very aggressive ones (Bellini's collecting duct carcinoma, Medullary carcinoma), and also that the sarcomatoid transformation, even in small areas, impacts the prognosis negatively. Childhood-characteristic renal carcinomas associated with chromosome translocations have been recognized (genetic fusion TFE3 or TFEB), as well as the family forms of renal carcinoma.
Regarding the UICC (International Union Against Cancer) classification, there are a series of aspects under argument (size, venous invasion, microvascular invasion, invasion of the adipous tissue of the renal sinus) that shall be discussed too, since it is possible that some modifications of the TNM might occur in the near future.
Key words: Renal cell carcinoma. Clear cell renal cell carcinoma. Papillary renal cell carcinoma. Chromophobe renal cell carcinoma. Bellíni´s carcinoma. Medulary renal cell carcinoma. New entities in renal cell carcinoma.
Las neoplasias que pueden surgir en el parénquima renal son múltiples y de muy variadas consecuencias para el paciente. Toda esta variedad ha sido recogida por la nueva clasificación de la OMS, pero como los carcinomas del parénquima renal son los más frecuentes en la edad adulta, representando un 6% de las neoplasias malignas esporádicas del adulto. En esta revisión nos limitaremos a ellos para valorar los nuevos aspectos clínicos que suponen las actuales clasificaciones.
Después de que Oberling y colaboradores al demostrar el origen tubular del carcinoma renal1 acabaran con la larga discusión sobre el origen de dicha neoplasia las clasificaciones internacionales unificaron a todos los tipos histológicos bajo la común denominación de adenocarcinoma renal, que podía ser de células claras o granulares, de arquitectura tubular, papilar o quística, y raramente de aspecto sarcomatoide2. Aunque parecía ya acabada la discusión con esta homogeneización, pronto empezaron a aparecer intentos de distinguir los subtipos histológicos según supuestos orígenes de distintos tramos de la nefrona, correlacionándolos con implicaciones clínicas diferentes.
Uno de los principales avances fue la descripción del Carcinoma renal cromófobo por Thoenes y cols3, de una morfología diferente al carcinoma de células claras y de probable origen en las células intercalares de la nefrona distal4. A partir de esta descripción (no aceptada inicialmente por las clasificaciones internacionales) se empezó a buscar con anticuerpos monoclonales la posibilidad de determinar distintos orígenes de los subtipos histológicos5; desafortunadamente no fue posible el poder definir grupos nítidos, por lo que no llegó a determinarse ningún tipo de modificación de la clasificación establecida.
No sería hasta la introducción de los estudios cromosómicos inicialmente de Kovacs6 y redefinidos posteriormente en la clasificación de Heildelberg7 que volverían a tenerse en cuenta los subtipos microscópicos del cáncer renal, ya que se comprobó que existía una correspondencia entre anomalías genéticas y fenotipo microscópico.
A partir de estas consideraciones se ha llegado a la última clasificación de la OMS en 20048, en donde se unen las características morfológicas, y genéticas, y se empiezan a reconocer ciertas variantes con evidencias de inmunofenotipos distintos o cambios genéticos y moleculares con implicaciones clínicas tanto por determinar diferentes agresividades como por abrir nuevas expectativas terapéuticas.
El principal criterio clasificatorio de la clasificación de la OMS es el aspecto celular y las diferentes variantes de carcinoma renal son expuestas de una manera consecutiva, sin embargo ya existen suficientes evidencias genéticas e inmunohistoquímicas como para poder adscribir los diferentes tipos a uno u otro segmento de la nefrona9 (Tabla 1). A pesar de ello se expondrán los subtipos tal como los ha establecido recientemente la OMS.

Carcinoma de células claras (Fig. 1)
Tal como su nombre indica esta neoplasia está constituida por células de citoplasma claro debido a su gran contenido en glucógeno y lípidos que se disuelven durante el procesamiento histológico. Ocasionalmente se pueden encontrar células con una mayor proporción de mitocondrias, lo que condiciona que adquieran un aspecto eosinófilo o granular, siendo excepcional que este tipo celular sea el predominante. Los núcleos son redondeados y sus características dependen del grado de diferenciación, que comporta diferencias biológicas y que será considerado más adelante en los factores pronósticos. La distribución de las células suele ser en masas sólidas con una estroma capilar muy abundante (que en ocasiones llega a formar lagos hemáticos) o tubular (ocasionalmente microquística)10, muy raramente se observan focos de tipo papilar11 y en un 5% de los casos es de predominio fusocelular (sarcomatoide)12.

La tumoración suele ser única en los casos esporádicos, con un 4% de multiplicidad y un 3% de bilateralidad. Está bien delimitada por una banda fibrosa (cápsula tumoral) consecuencia de la compresión de los tejidos de alrededor. Por las características citoplasmáticas la superficie de corte es de color amarillo (por los lípidos) y con áreas de hemorrágicas. Ocasionalmente hay áreas cicatriciales, e incluso con calcificación. La necrosis se relaciona con las neoplasias más agresivas.
El aspecto quístico que en ocasiones tiene puede ser por necrosis (pseudoquistes) o por estar constituida por verdaderos quistes neoplásicos, que pueden llegar a proporcionar un aspecto macroscópico que obliga a diferenciarlo del quiste multilocular, y que por su excelente evolución se ha considerado en la clasificación de la OMS como una forma clínico patológica distinta y se le ha denominado Carcinoma renal quístico multilocular.
El perfil inmunohistoquímico se caracteriza por la expresión mucho más frecuente de citoqueratinas de bajo peso molecular (CAM 5.2 alrededor del 60%) que de alto peso molecular (CK14 un 3,7%)13. La vimentina se expresa en un 82,6%14. El CD10 en el 94% y el anticuerpo monoclonal RCC en el 85%15.
Cambios genéticos y moleculares
La pérdida alélica 3p (LOH 3p) es la anomalía genética más típica de este carcinoma presente en un 75,8% de los casos16, pero no es exclusiva de él17. En el brazo corto del cromosoma 3 se han localizado tres genes posiblemente involucrados con el carcinoma renal. El gen supresor en 3p25-26 (VHL) que coincide con el de la enfermedad de von Hippel Lindau, pero que se expresa en el 34-56% de los carcinomas esporádicos18; y los localizados en 3p14.2 (posible gen FHIT) y en 3p.12 cuya pérdida es más frecuentes que la del primero8.
La LOH 3p interfiere en la vía de la hipoxia celular que activa al VEGF y al PDGF que tiene un papel esencial en la angiogénesis, el transporte de la glucosa, la proliferación celular, la migración celular y la apoptosis y ayuda a la adaptación a la hipoxia del carcinoma de células claras19, todo ello abre la posibilidad de nuevos tratamientos para frenar los procesos que favorecen el crecimiento y metástasis del carcinoma.
Características epidemiológicas y clínicas específicas
Es la variante más frecuente de carcinoma renal en el adulto, representan un 66.8% de nuestros casos (en la literatura del 70 al 85%)20,21. Las formas esporádicas se presentan en la sexta década de la vida (edad media global de 59,4 años, para ambos sexos) y es tres veces más frecuente en varones. En las formas familiares se suele hallar en la enfermedad de von Hippel Lindau, como se indicará más adelante. La sintomatología es la común a todas las neoplasias renales, y en el momento actual gran parte de los pacientes son asintomáticos por ser hallazgos en exploraciones por otras causas. Cualquier tumor de células claras es considerado como carcinoma sea cual sea su tamaño.
Carcinoma renal papilar (Fig. 2)
A pesar de que el criterio de clasificación de los carcinomas renales se basa en el tipo celular, el carcinoma papilar renal se define por la distribución de sus células alrededor de ejes capilares (papilas) al menos en un 50 a 70% del tumor20, diferenciándose así de las ocasionales áreas papilares que pueden hallarse en otros tipos de carcinoma renal11. Al analizar los tipos celulares que recubren a dichas papilas, en un 73% son células basófilas (tipo 1), y un 42% de células eosinófilas (tipo 2)22,23; e incluso hay casos con patrón sólido que recuerdan papilas comprimidas24. Del 2 al 5% de estos tumores son fusocelulares18,37. Estas variaciones citológicas y arquitectónicas, tienen implicaciones pronosticas, ya que los tipo 1 (generalmente de bajo grado) son muy poco agresivos en contra de lo que sucede en los tipo 2 (de alto grado) y las variantes sarcomatoides22. La estroma puede ser fibrosa o con aspecto edematoso, y característicamente se pueden hallar macrófagos espumosos, sobretodo en el tipo 1. Son frecuentes la hemorragia, la necrosis y los cristales de colesterol20.
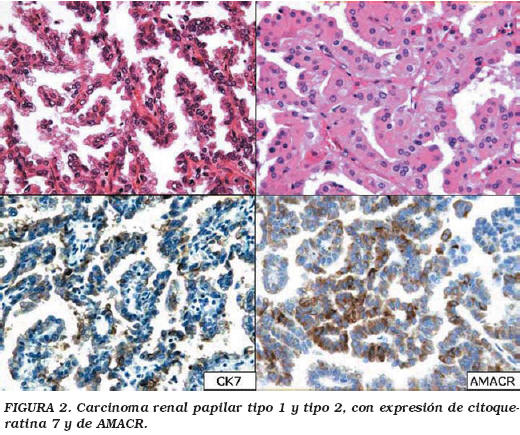
Se suelen expresar las citoqueratinas de bajo peso molecular: CAM 5.2 en casi todos los tumores y CK7 en un 75% de todos los casos siendo más frecuente en los tipo 1 que en los tipo 2 (87% versus 20%)25. La vimentina se expresa en un 85%14. El CD10 del 63 al 93%15,26. El anticuerpo monoclonal RCC en el 93%15. El EMA (MUC-1) del 40 al 60% de todos los casos27, siendo más frecuente en el tipo 1 que en el tipo 2 (72% versus 16.6%)28. Recientemente se ha encontrado que la racemasa alfa-metil-acil coenzima-A (AMACR-p504S) se expresa en todos los casos29.
Suelen ser tumores bien delimitados y al corte se observan con cierta frecuencia áreas necróticas y hemorrágicas. El aspecto es granular y rosado y la consistencia más blanda que en el resto de carcinomas. Suele ser más multifocal, incluso en los casos esporádicos, que los carcinomas de células claras y no es infrecuente el hallar pequeñas tumoraciones milimétricas (no mayores de 5 mm.) que se consideran microadenomas papilares.
Cambios genéticos y moleculares
Los cambios más frecuentes son la trisomía o tetrasomía 7, trisomía 17 y pérdida del cromosoma Y30. Estas alteraciones se han relacionado con la activación del proto-onco-gen c-MET (7q34), que codifica al HGFr31, que tanto pueden hallarse en las formas esporádicas como en las familiares, que ya serán consideradas más adelante. También se han detectado LOH 3p en un 59% de los casos siendo más frecuentes las LOH 3p25-26 (VHL) presentes en un 53´8% que las LOH 3p14.2 (FHIT) observadas en un 40.7%17. Otras trisomías en 12, 16 y 20 se consideran relacionadas con la progresión del tumor30.
En recientes estudios se ha encontrado que las alteraciones en 17q son casi exclusivas del tipo 1, mientras que las de 9p del tipo 2, lo que sugiere que cada uno de estos tipos celulares puede surgir de diferentes vías moleculares32.
Características epidemiológicas
Un 20% de nuestros casos de carcinoma renal son de tipo papilar (en la literatura oscila del 7 al 15%)20, la edad de presentación de las formas esporádicas es la misma que para el carcinoma de células claras (edad media de 59.7 años, similar en ambos sexos) y es siete veces más frecuente en el varón. Sólo los tumores de 5 mm. o menos se consideran totalmente benignos.
Carcinoma renal de células cromófobas (Fig. 3)
Las características que diferencian las células cromófobas de las células claras descritas son el mayor tamaño celular, el aspecto poliédrico con buena delimitación de la membrana citoplasmática (que le dan un aspecto de célula vegetal) y el abundante citoplasma reticular pálido por la presencia de vesículas que se tiñen con el hierro coloidal (tinción de Hale). Esta típica célula puede tener un citoplasma relativamente claro (subtipo de células claras) (pero nunca tanto como el del carcinoma de células claras) o más eosinófilo (variante eosinófila), dependiendo de la cantidad de mitocondrias33, existen formas intermedias que suponen una dificultad en el momento del diagnóstico34. En un 8,7% de los casos hay áreas sarcomatoides12. Por todas estas características sugieren que su origen es la célula intercalar de la nefrona distal.
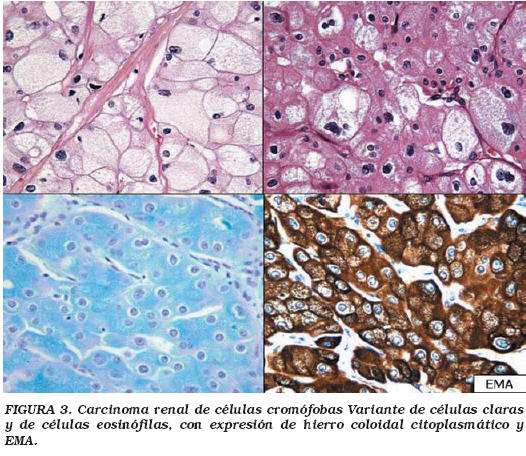
Suelen ser tumores de color pardo y ocasionalmente tienen una cicatriz central, lo que les asemeja al oncocitoma. Las formas esporádicas suelen ser únicas, bien delimitadas, las familiares suelen ser múltiples.
Tanto las citoqueratinas de bajo peso molecular como las de alto peso están presentes casi siempre13,35. Otros marcadores que suelen ser positivos son: El EMA (MUC-1)27,36 y el c-kit (88%)37. El CD10 suele ser negativo15, pero en algunas series llega a observarse hasta en un 26% de casos, que suelen corresponder a las formas más agresivas26.
Cambios moleculares y genéticos
Es típica las pérdidas en los cromosomas 1, 2, 6, 10, 13, 17 y 2138. La LOH 17 interrelaciona este tipo tumoral con el síndrome de Birt-Hogg-Dubé39.
También se han encontrado LOH 3p en un 86´6%, siendo algo más frecuentes las LHO 3p25-26 (71,4%) que las LOH 3p14.2 (66,6%)16.
En un 27% se casos hay mutación del TP5340 y también se han visto pérdidas alrededor del PTEN41.
Características epidemiológicas
Representan un 7,3% de nuestros casos (en la literatura se refiere entre el 5 y el 10%)42, La edad de presentación es similar a la de todos los carcinomas renales para ambos sexos. No existe predominio por sexos.
Por las características descritas, tanto microscópicas como macroscópicas se puede explicar la dificultad que en ocasiones supone el separar este carcinoma de un oncocitoma, y ésta circunstancia aún se explica más si como veremos más adelante hay formas híbridas y ciertas características genéticas que comparte con el oncocitoma.
Carcinoma de los conductos colectores de Bellini (Fig. 4)
Los criterios son imprecisos42, por ello la OMS ha definido criterios mayores y menores43. Son criterios mayores el alto grado nuclear de las células, el citoplasma eosinófilo, el patrón tubular acodado y la presencia de células en "tachuela de bota"44, con marcada desmoplasia y en ausencia de carcinoma urotelial; la presencia de carcinoma "in situ" en ductos de Bellini próximos al carcinoma es un criterio menor. El patrón sarcomatoide puede estar presente en un 29% de casos45.
Los estudios del inmunofenotipo son escasos, por lo que el porcentaje de casos que expresan diferentes anticuerpos son relativos. Teniendo en cuenta este hecho se ha observado expresión citoqueratinas de alto peso molecular (34bE12) y del Ulex europaeus en casi todos los casos46 lo que justifica que sean criterios mayores de la clasificación de la OMS43. La vimentina se expresa en un 100%. La CK7 y el EMA en un 33%47. Son negativas el CD1047 la AMACR48.

Las características microscópicas explican el aspecto macroscópico, suelen ser tumores de localización medular cuando son pequeños, de contornos mal definidos y aspecto infiltrativo, de coloración blanca y superficie fibrilar.
Cambios genéticos y moleculares
Hay muy pocos estudios citogenéticas. Las alteraciones más frecuentes son las pérdidas en los cromosomas 1q, 6p, 13q, 14, 15, 21q y 2249. Se ha descrito amplificación del HER2/ neu50.
Características epidemiológicas
Es muy difícil conocer la verdadera incidencia de esta neoplasia porque la mayoría de las veces es un diagnóstico de exclusión y se superpone al carcinoma urotelial, hecho que tanto puede ser expresión de un común proceso de carcinogénesis51 (tanto el urotelio como los ductos de Bellini son de origen wollfiano) como consecuencia puede haber confusión de ambas neoplasias en un solo grupo, por lo que algunos autores piden criterios aún más estrictos para el diagnóstico de carcinoma de ductos colectores52.
Carcinoma renal medular
Un crecimiento celular reticular y adenoide-quístico suele ser el patrón habitual de esta neoplasia, con células de citoplasma claro y eosinófilo de núcleos grotescos53,54. Se pueden encontrar áreas sarcomatoides55. La estroma característicamente es edematosa, con áreas de desmoplasia e inflamación aguda y crónica53,55.
Hay pocos estudios del inmunofenotipo, pero todos ellos coinciden en la expresión habitual de queratinas de bajo peso molecular (CAM 5.2) y la negatividad de las de alto peso (34bE12)55,56, así como del EMA (MUC-1)53,55,56. El Ulex europaeus se expresa focalmente en un 50% de los casos55.
El aspecto macroscópico refleja su gran agresividad al estar mal delimitado, localizado en la medular, blanco y fibrilar53,54,55. Curiosamente el riñón derecho se ha referido afectado tres veces más que el izquierdo54.
Cambios genéticos y moleculares
Es muy escasa la información sobre las alteraciones genéticas de este tumor, y los hallazgos comunicados oscilan desde la normalidad a la ocasional translocation t(3;8)(p21;q24)56 o monosomía del cromosoma 11 (el gen de la beta-globina se encuentra en 11p)57. Por los estudios moleculares, algunos autores consideran este tumor como una variante agresiva del carcinoma de ductos colectores de Bellini58. El HER2/neu es negativo y el VEGF y el HIF son extensamente positivos55.
Características epidemiológicas
Se describió por vez primera en 199553, desde entonces se han publicado alrededor de cien casos, todos ellos en gente joven (de 5 a 40 años), con casi el doble de casos en varones que en mujeres, la mayoría de ellos de raza negra con anemia falciforme54,55.
La sintomatología es la común a las neoplasias renales, aunque hay casos que simulan un proceso inflamatorio55. La mortalidad está cerca del 100% a los pocos meses después del diagnóstico53,54,55.
Carcinoma mucinoso tubular y fusocelular (Fig. 5)
Este subtipo ha sido incluido recientemente en la clasificación de la OMS, después de que diversos estudios habían llamado la atención de que ciertas neoplasias con células basófilas y túbulos elongados tenían un comportamiento clínico diferencial59.
Se caracteriza por células cuboides con escaso citoplasma, distribuidas en túbulos alargados, entremezcladas con zonas sólidas de células fusiformes de núcleos anodinos (no sarcomatoides). La estroma es edematosa con mucina extracelular60. Pueden verse macrófagos espumosos. Son tumores bien delimitados, de 3 a 10 cm, con un aspecto homogéneo de color amarillo-pardo.

Expresan mucho más frecuentemente la CK7 (100%) que las citoqueratinas de alto peso molecular (34bE12- 33%)61. La vimentina se expresa en todos los casos61. En 12 de nuestros pacientes con este tumor el CD10 y el c-kit son siempre negativos, el EMA (MUC-1) se expresa en todos los pacientes, pero es muy extenso sólo en el 50%. Cabe destacar que la AMACR está presente en el 92% de ellos. El Ulex europeaus es negativo62.
Cambios genéticos y moleculares
Las principales alteraciones cromosómicas son monosomías en 1, 4, 6, 8, 9, 13, 14, 15 y 2263, que junto a las características inmunohistoquímicas mencionadas apoyan el origen en la nefrona distal, habiéndose especulado sobre su posible origen en asa de Henle, pero no hay suficientes pruebas para confirmarlo64.
Características epidemiológicas
Los casos publicados son escasos, el predominio en mujeres es destacable. La edad media es de unos 55 años y la sintomatología es la común a las neoplasias renales, aunque se ha llamado la atención de su asociación con litiasis renal60. Tienen un excelente pronóstico64 incluso en algún caso con metástasis ganglionar local60.
Carcinomas renal inclasificable
En todo intento de clasificación histológica siempre existen casos concretos que son difíciles de adscribir a uno de los grupos, y esta situación ha llevado a que en la clasificación de la OMS se acepte un grupo de inclasificables
La incidencia de carcinomas renales no clasificados oscila entre el 3 y el 5%65 y es un grupo muy heterogéneo, pero su existencia permite la homogeneidad de los otros subtipos descritos hasta ahora y por lo tanto que los estudios tanto de incidencia, biología como de eventuales tratamientos sean mucho más precisos.
En nuestra opinión pueden ordenarse en tres apartados según la razón por la que son no clasificables.
Carcinomas renales anaplásicos o sarcomatoides puros (Fig. 6)
Para poder incluir un carcinoma renal en este subgrupo de inclasificados es obligado el haber tomado abundantes muestras representativas de todo el tumor, porque como ya se ha dicho todos las variantes pueden llegar a desarrollar formas sarcomatoides. Suelen ser localmente infiltrantes (42%), y ya metastáticos en casi un 50% de los casos, con una sobrevida media de 4,3 meses65.
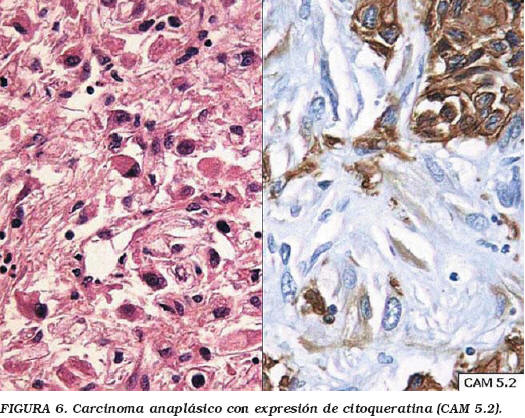
Dentro de este mismo grupo pueden incluirse los casos de carcinoma renal con características rabdoides (grandes células con glóbulos hialinos paranucleares) que no tienen nada que ver con los tumores renales rabdoides, y que se consideran como una divergencia clonal sobretodo de los carcinomas de células claras y de células cromófobas66,67, todos ellos con una gran agresividad biológica68.
Carcinomas (tumores) híbridos
Otra de las situaciones que conduce a que el patólogo considere que está frente a un carcinoma inclasificado es cuando se le mezclan en un mismo tumor distintos tipos celulares. Las formas híbridas más comunes son las producidas por el solapamiento de diferentes formas de tumores procedentes de la nefrona distal, sobretodo compuestos por oncocitoma + carcinoma renal de células cromófobas69 que son relativamente frecuentes en el síndrome de Birt.Hogg-Dubé70, pero que también hay descritas otras formas híbridas71,72,73.
Estos casos han de ser interpretados con mucho cuidado, ya que pueden confundirse patrones y tipos celulares, pero sí que parecen existir verdaderas formas de transición, justificables por las alteraciones genéticas, tal como ocurre en el síndrome de Birt-Hogg-Dubé.
El pronóstico de estos tumores es tema de discusión, aunque, aparentemente, el pronóstico de los tumores híbridos más frecuentes (oncocitoma +Carcinoma de células cromófobas) es el que corresponde al oncocitoma69.
Subtipos morfológicos no reconocidos por la OMS
A pesar de la progresiva incorporación de diferentes variantes a la clasificación de la OMS es tal la versatilidad morfológica del carcinoma renal que diferentes autores van publicando formas morfológicas no totalmente atribuibles a uno de los grupos descritos74,75. Estos casos contribuyen a la discusión y son el origen de posibles nuevas entidades, pero mientras no se llega a un consenso deben ser consideradas como inclasificables.
De entre estas variantes morfológicas quizás una de las más interesantes, por su buen pronóstico y porque representa una ampliación conceptual de un subtipo ya aceptado, es el Carcinoma de bajo grado de ductos colectores75. Esta neoplasma ya había sido descrita con la denominación de carcinoma túbulo-quístico de bajo grado76, se caracteriza por una proliferación de células neoplásicas de bajo grado, en túbulos, con mayor o menor dilatación, que pueden llegar a proporcionar un aspecto multiquístico75, sin componente fusocelular76 y con inmunofenotipo de nefrona distal61,76.
Carcinomas renales en la infancia y la adolescencia
En la infancia el carcinoma renal sólo representa el 4% de los tumores renales. En la literatura más reciente han ido apareciendo publicaciones refiriéndose a carcinomas renales en niños y adolescentes, con hallazgos a veces contradictorios ya que mientras algunos autores hallan una mayor incidencia de carcinoma renal papilar77, otros encuentran mayor proporción de carcinoma renal de células cromófobas78. Estas discrepancias están ocasionadas tanto por la escasez de casos como por la existencia de ciertas variantes específicas en estas edades tales como los carcinomas renales asociados con translocaciones cromosómicas79 que pueden tener células claras y eosinófilas y patrones de crecimiento tubulares y papilares.
En una reciente publicación de 41 casos80 se han encontrado un 14% de carcinomas de células claras, 22% de carcinomas renales papilares (con similar incidencia tanto en el tipo 1 como en el 2), 5% carcinomas de células cromófobas, 5% carcinomas de ductos colectores de Bellini, 7% de carcinomas a partir de un nefroblastoma, 2% de carcinomas asociados a neuroblastoma, y un 19,5% carcinomas de translocación. El 24% no se pudieron clasificar. Así pues se observa que cualquier tipo de tumor puede aparecer en estas edades, siendo específico de ellas los tumores de translocación.
Carcinomas renales asociados a translocaciones cromosómicas (fusión genética TFE3 o TFEB)
Las células de estos carcinomas pueden ser claras y eosinófilas con amplios y voluminosos citoplasmas que le dan un aspecto de pompas de jabón, asimismo se pueden encontrar cuerpos de psamoma calcificados. Una peculiaridad que ayuda también al diagnosticó es el encontrar casi siempre un patrón papilar asociado a los otros patrones81. Según el tipo de translocación parece que pueden haber sutiles diferencias microscópicas en el volumen de las células80,82.
Cambios genéticos
Pueden haber diversas alteraciones genéticas en Xp11.2, pero todas ellas producen fusiones en el gen TFE3, las descritas son: t(X;1)(p11,2;q21) fusión PRCC y TFE3 (es la más frecuente); t(X;17) (p11.2; q25) fusión ASPL y TFE3; t(X;1) (p11.2;p34) fusión PSF y TFE3; Inv(X)(p11;q12) fusión NoO(p54nrb) y TFE3100. Otra translocación es la t(6;11)(p21;q12) fusión TFEB83
La CAM 5.2 y la CK7 se expresan en un 45% de casos, de forma focal. La vimentina se expresa en un 63%, el CD10 y el anticuerpo monoclonal RCC en el 100%, el EMA (MUC-1) en el 45%81. Como consecuencia de la translocación la fusión genética codifica una nueva proteína quimérica que retiene al factor de transcripción TFE3 o el TFEB y podemos detectarlos con inmunohistoquímica en los núcleos de las células neoplásicas, mientras que es extremadamente rara su positividad en los tejidos normales82.
Características epidemiológicas
Estas neoplasias son propias de la segunda y tercera década de la vida, más frecuentes en el sexo femenino y no se tienen suficientes datos para conocer su verdadera biología79,81.
Factores anatomo-patológicos pronósticos
Subtipos histológicos
En la nueva clasificación de la OMS queda muy claro que ciertos subtipos de tumor tienen un excelente pronóstico (Carcinoma renal quístico multilocular, Carcinoma papilar renal de tipo 1, Carcinoma mucinoso tubular y fusocelular), otros son muy agresivos (Carcinoma de ductos colectores de Bellini, Carcinoma medular) y que la transformación sarcomatoide, aún en pequeñas áreas, tiene un impacto negativo sobre el pronóstico45.
Las diferencias pronósticas de las formas más habituales (menor agresividad del carcinoma renal de células cromófobas y mayor agresividad del carcinoma de células claras) son estadísticamente significativas en los estudios univariantes84,85, pero en algún otro estudio no tiene un significado independiente si se tienen en cuenta los otros factores pronósticos tales como grado y el estadio21. A pesar de ello sí que estos subtipos celulares parecen relacionarse con biologías distintas (86% de los carcinomas de células cromófobas son de bajo grado y están localizados al riñón)86.
Otro aspecto que no debe menospreciarse es la percepción de que las respuestas a las terapéuticas post-nefrectomía son distintas87 y ello ya está llevando a ser muy estrictos en la inclusión de casos en estudios terapéuticos; por otra parte las diferencias genéticas y moleculares están poniendo de manifiesto nuevas vías farmacológicas.
Grado de diferenciación nuclear
A pesar de las críticas sobre la reproducibilidad del grado nuclear de Fuhrman (basado en el tamaño y forma del núcleo, así como en la presencia o no de nucleolo) éste sigue siendo un buen parámetro, y así se han comunicado sobrevidas cáncer específicas a 5 años del 89%, 65% y 46% para los grados 1, 2 y 3-488. Actualmente se está recomendando el establecer el grado de diferenciación en todos los subtipos celulares y no sólo en los de células claras y se tiende a subdividirlos en bajo grado (grado 1 y 2) y alto grado (grados 3 y 4)89.
Estadio anatómico
El TNM 2002 (Tabla 2) tiene una utilidad global muy aceptable.

Los aspectos más controvertidos son:
El tamaño tumoral. El nivel de corte a 7 cm para los T1 está discutido90,91,92, y parece ser que sería más significativo que fuera de 5 cm114. Esta controversia denota la importancia pronóstica del tamaño, pero no ha de influir en la decisión de la cirugía conservadora del riñón93.
Trombo tumoral en vena. En ausencia de invasión local por contigüidad no parece acarrear tan mal pronóstico como se había supuesto (sobrevida a 5 años del 47 al 69%)94, y la invasión de la vena cava inferior no parece tener repercusión en la sobrevidada95.
Invasión suprarrenal. La sobrevida a 5 años de los pacientes con invasión del tejido adiposo es del 35%, y aquellos con extensión por contigüidad a la adrenal del 0%96 lo que induce a un cambio en la valoración del TNM actual.
Invasión de la grasa del seno renal. La invasión del tejido adiposo del seno parece ser la forma más frecuente de invasión del tejido adiposo y tener una mayor agresividad, probablemente por la abundancia de vasos venosos. Se correlaciona con el grado de diferenciación y el tamaño del tumor97.
Todos estos datos confirman la necesidad de un preciso estudio de los especimenes quirúrgicos, siguiendo las normativas que se van publicando en las diferentes sociedades98,99.
Otros factores que se están estudiando para poder incorporar a los ya dichos son la NECROSIS que se relaciona con el pronostico en los carcinomas de células claras y de células cromófobas pero no en los carcinomas renales papilares85 y la invasión de vasos del parénquima renal de alrededor del tumor100.
Tumores renales familiares
Casi todas las variantes descritas pueden expresarse en formas familiares, llegando a representar un 4% de todos los carcinomas renales9. La mayoría de las veces son múltiples, bilaterales y aparecen en edades más tempranas que sus correspondientes formas esporádicas. Muchos de ellos están en el contexto de neoplasias en otras localizaciones.
Las formas más importantes hasta ahora reconocidas son101:
Enfermedad de Von Hippel-Lindau (VHL)
Autosómica dominante. El gen de VHL está localizado en el cromosoma 3p25. Se asocian el hemangioma de retina, carcinoma de células claras de riñón, hemangioblastoma del cerebelo y en médula espinal, feocromocitoma, tumores endocrinos de páncreas y cistadenoma del epidídimo102. La edad media de expresión del carcinoma renal es de 37 años, la causa principal de fallecimiento son las metástasis del carcinoma renal.
Carcinoma renal papilar hereditario
Este síndrome se caracteriza por la presencia de carcinomas papilares tipo 1. La presencia de abundantes tumores de muy diversos tamaños, en ambos riñones, debe hacernos sospechar de este tipo de síndrome. Aunque suele predominar la afectación renal aislada, también se han comunicado tumores de mama, páncreas, pulmón, piel y estómago.
Complejo de la esclerosis tuberosa (TSC)
Forma autosómica dominante con afectación muy amplia de distintos tejidos con angiofibromas faciales, fibromas sububgueales, fibromas cutáneos planos, máculas hiperpigmentadas, angiomiolipomas renales y carcinomas de células claras (estos últimos ocasionalmente)103. Se relacionan con dos genes el TSC1 (9q34), TSC2 (16p13.3).
El síndrome de Birt-Hogg-Dubé (BHD)
Es un síndrome autosómico dominante con una tríada clásica de múltiples lesiones cutáneas (fibrofoliculomas, tricodiscomas y acrocordones) y tumores renales múltiples y bilaterales, principalmente oncocitomas o carcinomas de células cromófobas, aunque también se han descrito de células claras y papilares39 también se ha asociado con carcinoma de colon, lipomas múltiples y neumotórax espontáneo. Su gen se localiza en 17q12-q11.2.
Conclusión
Ante lo expuesto se puede concluir que la nueva clasificación de la OMS (2004) para los carcinomas renales nos ayuda a estratificar los carcinomas en grupos de distintas agresividades, permite hacer grupos histológicos homogéneos al poder considerar las formas peculiares como inclasificables, y facilita la uniformidad de las series en ensayos clínicos. La clasificación de la UICC continúa siendo útil, pero probablemente en la revisión del 2007 se harán ciertas matizaciones para poder establecer mejores grupos de riesgo. Por lo que la generalización del uso de las clasificaciones internacionales, con el máximo de escrupulosidad, nos permite una mejor categorización pronóstica y terapéutica de utilidad para el enfermo.
Referencias
1. Oberling C, Rivière M, Haguenan F.: Ultrastructure of the clear cells in renal carcinomas and its importance for the demonstration of their renal origin. Nature. 1960;186:402-403 [ Links ]
2. Bennington JL, Beckwith JB.: Tumors of the kidney, renal pelvis, and ureter. Atlas of tumor pathology. Second series. Fascicle 12 p-130. AFIP Washington 1975. [ Links ]
3. Thoenes W, Störtkel S, Rumpelt HJ.: Human chromophobe cell renal carcinoma. Wirchows Arch (Cell Pathol) 1985; 155:277-287. [ Links ]
4. Störkel S, Pannen B. Thoenes W, Staert PV, Wagner S, Drenckhalm D.: Intercalated cells as a probable source for the development of renal oncocitoma. Wirchows Arch (B). 1988;56:185-189. [ Links ]
5. Liu GF, Song-Liang C, Bi-Juan C, Chieh-Ping W.: Cellular origin of renal cell carcinoma. An immunohistochemical study on monoclonal antibodies. Scand J Urol Nephrol 1991;38(S):203-206. [ Links ]
6. Kovacs G.: Molecular differential pathology of renal cell tumours . Histopathology 1993;22 (1):1-8. [ Links ]
7. Kovacs G, Akhtar M, Beckwith BJ.: The Heidelberg classification of renal cell tumours. J. Pathol. 1997;183:131-133. [ Links ]
8. Eble JN, Sauter G, Epstein JI, Sesterhenn IA.: World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p.-10, IARC Press, Lyon 2004. [ Links ]
9. Odmer D, van den Hurk W, van Groningen JJ, Eleveld MJ, Martens GJ, Weterman MA, van Kessel AG.: Understanding familial and non-familial renal cell cancer. Hum Mol Genet. 2002;11:2489-2498. [ Links ]
10. Grignon DH, Eble JN, Bonsib SM, Moch H.: Clear cell renal cell carcinoma In Eble JN, Sauter G, Epstein JI, Sesterhenn IA World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p.-23, IARC Press, Lyon 2004. [ Links ]
11. Füzesi L, Gunawan B, Bergmann F, Tack S, Braun S, Jakse G.: Papillary renal cell carcinoma with clear cytomorphology and chromosomal loss of 3p. Histopathology 1999;35(2):157-161. [ Links ]
12. Cheville JC, Lohse CM, Zincke H, Weaver AL, Leibovich BC, Frank I, Blute ML.: Sarcomatoid renal cell carcinoma. An examination of underluing histologic subtype and analysis of associations with patient outcome. Am J Surg Pathol. 2004;28(4):435-441. [ Links ]
13. Chu PG, Weiss LM.: Cytokeratin 14 immunoreactivity distinguishes oncocytic tumour from its renal mimics: an immunohistochemical study of 63 cases. Histopathology 2001;39(5):455-462. [ Links ]
14. Young AN, de Oliveira Sales PG, Lim SD, Cohen C, Petros JA, Marshall FF, Neish AS, Amin MB.: Beta-Defensin-1, Parvalbumin and vimentin. A panel of diagnostic immunohistochemical markers for renal tumors derived from gene expression profiling studies using cDNA microarrays. Am J Surg Pathol 2003;27(2):199-205. [ Links ]
15. Avery AK, Beckstead J, Renshaw AA, Corless CL.: Use of antibodies to RCC and CD10 in the differential diagnosis of renal neoplasms. Am J Surg Pathol 2000;24(2):203-210. [ Links ]
16. Velickovic M, Delahunt B, Grebe SKG.: Loss of heterozygosity at 3p14.2 in clear cell carcinoma is an early event and is localized to the FHIT gene locus. Cancer Res 1999;59(6): 1323-1326. [ Links ]
17. Velickovic M, Delahunt B, Störkel S, Grebe SKG.: VHL and FHIT locus loss of heterozygosity is common in all renal cancer morphotypes but differs in pattern and prognostic significance. Cancer Res 2001;61(12):4815-4819. [ Links ]
18. Schraml P, Struckmann K, Hatz F, Sonnet S, Kully C, Gasser T, Sauter G, Mihatsch MJ, Moch H.: VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J Pathol 2002;196(2);186-193. [ Links ]
19. Lam JS, Shvarts O, Leppert JT, Figlin RA, Belldegrun AS.: Renal cell carcinoma 2005: New frontiers in staging, prognostication and targeted molecular therapy. J Urol 2005; 173(6):1853-1862. [ Links ]
20. Fleming S, O´Donell M.: Surgical pathology of renal epithelial neoplasms: recent advances and current status. Histopathology 2000;36(3):195-202. [ Links ]
21. Patard JJ, Leray E, Rioux-Leclercq N, Cindolo L, Ficarra V, et al. Prognostic value of histologic subtypes in renal cell carcinomas: a multicenter experience. J Clin Oncol 2005;23:2763-2771. [ Links ]
22. Delahunt B, Eble JN.: Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997;10(6):537-544. [ Links ]
23. Amin MB, Corless CL, Renshaw AA, Tickoo SK, Kubus J, Schultz DS.: Papillary (Chromophil) renal cell carcinoma: Histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 62 cases. Am J Surg Pathol 1997;21:621-635. [ Links ]
24. Renshaw AA, Zhang H, Corless CL, Fletcher JA, Pins MR.: Solid variants of papillary (Chromophil) renal cell carcinoma: Clinicopathologic and genetic features. Am J Surg Pathol 1997;21:1203-1209. [ Links ]
25. Delahunt B, Eble JN.: Papillary renal cell carcinoma. In Eble JN, Sauter G, Epstein JI, Sesterhenn IA World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p.-27, IARC Press, Lyon 2004. [ Links ]
26. Martignoni G, Pea M, Brunelli M, Chilosi M, Zamó A, Bertaso M, Cossu-Rocca p, Eble JN, Mikuz G, Puppa G, Badonal C, Ficarra V, Novella G, Bonetti F.: CD10 is expressed in a subset of chromophobe renal cell carcinomas. Mod Pathol 2004;17(12):1455-1463. [ Links ]
27. Langner C, Ratschek M, Rehak P, Schips L, Zigeuner R.: Expression of MUC1(EMA) and E-cadherin in renal cell carcinoma: a systematic immunohistochemical analysis of 188 cases. Mod Pathol 2004;17(2):180-188. [ Links ]
28. Leroy X, Zini L, Leteurtre E, Zerimech F, Porchet N, Aubert JP, Gosselin B, Copin MC.: Morphologic subtyping of papillary renal cell carcinoma: Correlation with prognosis and differential expression of MUC1 between the two subtypes. Mod Pathol 2002;15(11):1126-1130. [ Links ]
29. Tretiakova MS, Sahoo S, Takahashi M, Turkyilmaz M, Vogelzang NJ, Lin F, Krausz T, The BT, Yang XJ.: Expression of alpha-methyllacil-CoARacemase in papillary renal cell carcinoma. Am. J. Surg. Pathol. 2004;28:69-76. [ Links ]
30. Kovacs G, Fucesi L, Emanual A, Kung HF.: Cytogenetics of papillary renal cell tumors. Genes Chromosomes Cancer. 1991;3:249-255. [ Links ]
31. Schmidt L, Junker K, Weirich G, Glenn G, Choyke P, Lubensky I, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58(8):1719-22. [ Links ]
32. Sanders ME, Mick R, Tomaszewski JE, Barr FG.: Unique patterns of allelic imbalance distinguish type 1 from type2 sporadic papillary renal cell carcinoma. Am J Pathol 2002;161:997-1005. [ Links ]
33. Thoenes W, Störkel S, Rumpelt H-J, Moll R, Baum HP, Werner S.: Chromophobe cell renal carcinoma and its variants- a report on 32 cases. J Pathol 1988;155:277-287. [ Links ]
34. Akhtar M, Kardar H, Linjawi T, McClintock J, Ali MA.: Chromophobe cell carcinoma of the kidney: a clinicopathologic study of 21 cases. Am J Surg Pathol 1995; 19(11):1245-1256. [ Links ]
35. Wu SL, Kothari P, Wheeler TM, Reese T, Connelly JH.; Cytokeratins 7 and 20 immunoreactivity in chromophobe renal cell carcinomas and renal oncocytomas. Mod Pathol 2002;15(7):712-717. [ Links ]
36. Abrahams NA, MacLennan GT, Khoury JD, Ormsby AH, Tamboli P, Doglioni C, Schumaker B, Tickoo SK.: Chromophobe renal cell carcinoma: a comparative study of histological, immunohistochemical and ultrastructural features using high throughput tissue microarray. Histopathology 2004;45:595-602. [ Links ]
37. Petit A, Castillo M, Santos M, Mellado B, Alcover J, Mallofré C.: kit expression in chromophobe cell carcinoma. Comparative immunohistochemical analysis of kit expression in different renal cell neoplasms. Am J Surg Pathol 2004;28(5):676-678. [ Links ]
38. Speicher MR, Schoell B, du Manoir S, Schrock E, Ried T, Cremer T, Störkel S, Kovacs G.: Specific loss of chromosomes 1, 2, 6, 10, 13, 17 and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol 1994; 145: 356-364. [ Links ]
39. Roth JS, Rabinowitz AD, Benson M, Grossman ME.: Bilateral renal cell carcinoma in the Birt-Hogg-Dubé syndrome Am Acad Dermatol. 1993;6:1055-1056. [ Links ]
40. Contractor H, Zariwala M, Bugert P, Zeisler J, Kovacs G.: Mutation of the p53 tumour suppressor gene occurs preferentially in the chromophobe type of renal cell tumour . J Pathol 1997;181:136-139. [ Links ]
41. Sukosd F, Digon B, Fischer J, Pietsch T, Kovacs G.: Allelic loss at 10q23.3, but lack of mutation of PTEN/MMAC1 in chromophobe renal cell carcinoma. Cancer Genet Cytogenet. 2001;128:161-153. [ Links ]
42. Renshaw AA,; Subclassification of renal neoplasms: an update for practicing pathologists. Histopathology 2002;41:283-300. [ Links ]
43. Srigley JR, Moch H.: Carcinoma of the collecting ducts of Bellini. In Eble JN, Sauter G, Epstein JI, Sesterhenn IA World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p.- 33, IARC Press, Lyon 2004. [ Links ]
44. Srigley JR, Eble JN.: Collecting duct carcinoma of kidney. Sem Diagn Pathol 1998;15:54-57. [ Links ]
45. De Peralta-Venturina M, Moch H, Amin M, Tamboli O, Hailemariam S, Mihatsch M, Javidan J, Stricker H, Ro JY, Amin MB.: Sarcomatoid differentiation in renal cell carcinoma. A study of 101 cases. Am J Surg Pathol 2001;25: 275-284. [ Links ]
46. Flening S, Symes CE.: The distribution of cytokeratin antigens in the kidney and in renal tumours. Histopathology 1987;11:157-170. [ Links ]
47. Mazal PR, Stichenwirth M, Koller A, Blach S, Haitel A, Susani M.: Expression of aquaporins and PAX-2 compared to CD10 and cytokeratin 7 in renal neoplasms: a tissue microarray study. Mod Pathol 2005;18(4):535-540. [ Links ]
48. Tretiakova MS, Sahoo S, Takahashin M, Turkyilmaz M, Vogelzang NJ, Lin F, Krausz T, Teh BT, Yang XJ.: Expression of alpha-methylacil-CoA Racemase in papillary renal cell carcinoma. Am J Surg Pathol 2004;28:69-76. [ Links ]
49. Füzesi L, Cober M, Mittermayer CH.: Collecting duct carcinoma: cytogenetic characterization. Histopathology 1992;21:155-160. [ Links ]
50. Selli C, Amorosi A, Vona G, Sestini R, Travaglini F, Bartoletti F, Orlando C.: Retrospective evaluation of cerbB- 2 oncogene amplification using competitive PCR in collecting duct carcinoma of the kidney. J Urol 1997; 158(1):245-247. [ Links ]
51. Orsola A, Trias I, Raventós CX, Español I, Cecchini L, Orsola I.: Renal collecting (Bellini) duct carcinoma displays similar characteristics to upper tract urothelial cell carcinoma. Urology 2005;65:49-54. [ Links ]
52. Kafe H, Verbavatz JM, Cochand-Priollet B, Castagnet P, Viellefond.: Collecting duct carcinoma: an entity to be redefined?. Virchows Arch. 2004; 445: 637-640. [ Links ]
53. Davis CJ, Mostofi FK, Sesterhenn IA.: Renal medullary carcinoma the seventh sickle cell nephropaty. Am J Surg Pathol 1995;19:1-11. [ Links ]
54. Dimashkieh H, Choe J, Mutema G.: Renal medullary carcinoma. A report of 2 cases and review of the literatura. Arch Pathol Lab Med 2003;127:135-138. [ Links ]
55. Swartz MA, Karth J, Schneider DT, Rodríguez R, Beckwith JB, Perlman EJ.: Renal medullary carcinoma, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology 2002;60:1083-1089. [ Links ]
56. Rodriguez-Jurado R, González-Crussi F. Renal medullary carcinoma. Immunohistochemical and ultrastructural observations. J Urol Pathol 1996;4:191-203. [ Links ]
57. Avery RA, Harris JE, Davis CJJr, Borgaonkar DS, Byrd JC, Weiss RB.: Renal medullary carcinmoma: clinical and therapeutic aspects of a newly described tumor. Cancer 1996;78:128-132. [ Links ]
58. Polascik TJ, Bostwick DG, Cairos P.: Molecular genetics and histopathologic features of adult distal nephron tumors. Urology 2002;60:941-946. [ Links ]
59. Srigley JR.: Mucinous tubular and spindle cell carcinoma In Eble JN, Sauter G, Epstein JI, Sesterhenn IA World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p-40,IARC Press, Lyon 2004. [ Links ]
60. Hes O, Hora M, Perez-Montiel DM, Suster S, Curik R, Sokol L, et al. Spindle and cuboidal renal cell carcinoma, a tumor having frequent association with nephrolitiasis: report of 11 cases including a case with hybrid conventional renal cell carcinoma/ spindle and cuboidal renal cell carcinoma components. Histopathology 2002;41:549-555. [ Links ]
61. Skinnider BF, Folpe AL, Hennigar RA, Lim SD, Cohen C, Tamboli P, Young A, Peralta-Venturina M, Amin MB.: Distribution of cytokeratins and vimentin in adult renal neoplasms and normal renal tissue. Potential utility of a cytokeratin antibody panel in the differential diagnosis of renal tumors. Am J Surg Pathol 2005;29:747-754. [ Links ]
62. Arwani AV, Husain AN, Epstein JI, Beckwith JB, Argani P.: Low-grade myxoid renal epithelial neoplasms with distal nephron differentiation. Hum Pathol 2001;32:506-512. [ Links ]
63. Rakozy C, Schmahl GE, Bogner S, Störkel S.: Low-grade tubular mucinous renal neoplasms: Morphologic, immunohistochemical, and genetic features. Mod Pathol 2002;15:1162-1171. [ Links ]
64. Weber A, Srigley J, Moch H.: Mucinous spindle cell carcinoma of the kidney. A molecular analysis. Pathologe 2003;24:453-459. [ Links ]
65. Zisman A, Chao DH, Pantuck AJ, Kim HJ, Wieder JA,Figlin RA, Said JW, Belldegrun AS.: Unclassified renal cell carcinoma: Clinical features and prognostic impact of anew histological subtypes. J Urol 2002;168(3):950-955. [ Links ]
66. Shannon B, Wisniewski ZS, Bentel J, Cohen RJ.: Adult rhabdoid renal cell carcinoma. Divergent differentiation of conventional (Clear cell) carcinoma. Arch Pathol Lab, Med 2002;126:1506-1510. [ Links ]
67. Shannon BA, Cohen RJ.: Rhabdoid differentiation of chromophobe renal cell carcinoma. Pathology 2003;35:228-230. [ Links ]
68. Gokden N, Nappi O, Swanson PE, Pfeifer JD, Vollmer RT, Wick MR, Humphrey PA.: Renal cell carcinoma with rabdoid features. Am J Surg Pathol 2000;24(10):1329-1338. [ Links ]
69. Tickoo sK, Reuter VE, Amin MB, Srigley JR, Epstein JI, Min KW, Rubin MA, Ro JY.: Renal oncocytosis. A morphologic study of fourteen cases. Am J Surg Pathol 1999;23(9):1094-1101. [ Links ]
70. Pavlovich CP, Walter MM, Eyler RA, Hewitt SM, Zbar B, Linehan WM, Merino MJ.: Renal tumors in the Birt-Hogg-Dube syndrome. Am. J. Surg. Pathol. 2002;26:1542-1552. [ Links ]
71. Lindgren V, Paner GP, Flanigan RC, Clark JI, Campbell SC, Picken MM.: Renal tumor with overlapping distal nephron morphology and karyotype. Arch Pathol Lab Med 2004;128:1274-1278. [ Links ]
72. Gong Y, Sun X, Haines GK, Pins MR.: Renal cell carcinoma, chromophobe type, with collecting duct carcinoma and sarcomatoid components. Arch Pathol Lab Med 2003;127:38-40. [ Links ]
73. Matei DV, Rocco B, Varela R, Verwei F, Scardino E, Renne G, De Cobelli O.: Synchronous collecting duct carcinoma and papillary renal cell carcinoma: A case report and review of the literature. Anticancer Res. 2005;25:579-586. [ Links ]
74. Amin MB, Michal M, Radhakrishnan A, Hes O, McKenney JK, Cheville JC.: Primary thyroid-like follicular carcinoma of the kidney: A histological distinctive primary renal epithelial tumor. Mod. Pathol. 2004; 17(S1):136A (A 567). [ Links ]
75. Farah R, Ben-Izhak O, Munichor M, Cohen H.: Low-grade renal collecting duct carcinoma. A case report with histochemical, immunohistochemical, and ultrastructural study. Ann Diagn Pathol 2005;9:46-48. [ Links ]
76. MacLennan GT, Farrow GM, Bostwick DG.: Low-grade collecting duct carcinoma of the kidney: report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology 1997;50:679-684. [ Links ]
77. Renshaw AA, Granter SR, Fletcher JA, Kozakewick HP, Corless CL, Perez-Atayde AR.: Renal cell carcinomas in children and young adults. Increased incidence of papillary architecture and unique subtypes. Am J Surg Pathol 1999;23(7):795-802. [ Links ]
78. Gillett MD, Cheville JC, Karnes RJ, Lohse CM, Kwon ED, Leibovich BC, Zincke H, Blute ML.: Comparison of presentation and outcome for patients 18 to 40 and 60 to 70 years old with solid renal masses. J Urol 2005;173(6): 1893-1896. [ Links ]
79. Argani P, Ladanyil M.: Renal carcinomas associated with Xp11.2 translocations/TFE3 gene fusions. In Eble JN, Sauter G, Epstein JI, Sesterhenn IA World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p.- 37, IARC Press, Lyon 2004. [ Links ]
80. Bruder E, Passera O, Harms D, Leuschner I, Ladany M, Argani P, Eble JN, Struckmann K, Schraml P, Moch H.: Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol 2004;28(9):1117-1132. [ Links ]
81. Argani P, Antonescu CR, Couturier J, Fournet JC, Sciot R, Debiec-Rychter M, et al. PRCC-TFE3 renal carcinomas. Morphologic, immunohistochemical, ultrastructural, and molecular analysis of an entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol2002;26(12):1553-1566. [ Links ]
82. Argani P, Lal P, Hutchinson B, Lui MY, Reuter VE, Ladanyi M. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions. A sensitive and specific immunohistochemical assay. Am J Surg Pathol 2003;27(6):750-761. [ Links ]
83. Kuiper RP, Schepens M, Thijssen J, van Asseldonk M, van den Berg E, Bridge J, Schuuring E, Schoenmakers EFPM, van Kessel AG.: Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum Mol Genet 2003;12:1661-1669. [ Links ]
84. Amin MB, Amin MB, Tamboli P, Javidan J, Stricker H, De- Peralta Venturina M, Deshpande A, Menon M.: Prognostic impact of histologic subtyping of adult renal epithelial neoplasms. An experience of 405 cases. Am J Surg Pathol 2002;26(3):281-291. [ Links ]
85. Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histological subtypes of renal cell carcinoma. Am J Surg Pathol 2003;27(5):612-624. [ Links ]
86. Renshaw AA, Richie JP.: Subtypes of renal carcinoma. Different onset and sites of metastatic disease. Am J Clin Pathol 1999;111(4):539-549. [ Links ]
87. Motzer RJ, Bacik J, Mariani T , Russo P, Mazumdar M, Reuter V: Treatment outcome and survival associated with metastatic renal cell carcinoma of non-clear-cell histology. J Clin Oncol 2002;20(9):2376-2381. [ Links ]
88. Tsui KH, Shvarts O, Smith RB, Figlin RA, deKernion JB, Belldegrun A.: Prognostic indicators for renal cell carcinoma: a multivariate analysis of 643 patients using the revised 1997 TNM staging criteria. J Urol 2000;163(4):1090-1095 [ Links ]
89. Algaba F.: Prognostic factors of epithelial tumours of the kidney. Pathologica 1999;91(1):51-53. [ Links ]
90. Ficarra V,Prayer-Galetti T, Novara G, Bratti E, Zanolla L, dak Bianco M, Artibani W, Pagano F.: Tumor-size breakpoint for prognostic stratification of localized renal cell carcinoma. Urology 2004;63(2):235-240. [ Links ]
91. Wunderlich H, Dreihaup M, Schlichter A, Kosmehl H, Reichelt O, Schubert J.: New cut-off point between T1 and T2 renal cell carcinoma-Necessary for a better discriminatory power of the TNM classification. Urol Int 2004;72(2):123-128. [ Links ]
92. Frabk I, Blute ML, Leibovich BC, Cheville JC, Lohse CM, Kwon ED, Zincke H.: pT2 classification for renal cell carcinoma. Can its accuracy be improved?. J Urol 2005;173(2):380-384. [ Links ]
93. Patard JJ, Shvarts O, Lam JS, Pantuck AJ, K8im HL, Ficarra V, Cindolo L, Han KR, De la Taille A, Tostain J, Artibani W, Abbou CC, Lobel B, Chopin DK, Figlin RA, Mulders PF, Belldegrun AS : Safety and efficacy of partial nephrectomy for all T1 tumors based on an international multicenter experience. J Urol 2004;171(6 pt1):2181-2185. [ Links ]
94. Zisman A, Wieder JA, Pantuck AJ, Chao DH, Dorey F, Said JW, Gitlitz BJ, deKernion JB, Figlin RA, Belldegrun AS.: Renal cell carcinoma with tumor thrombus extension: biolog, role of nephrectomy and response to immunotherapy. J Urol 2003;169(3):909-916. [ Links ]
95. Moinzadeh A, Libertino JA.: Prognostic significance of tumor thrombus level in patients with renal cell carcinoma and venous tumor thrombus extension. Is all T3b the same?. J Urol 2004;171(2pt1):598-601. [ Links ]
96. Lam JS, Shvarts O, Leppert JT, Figlin RA, Belldegrun AS.: Renal cell carcinoma 2005: new frontiers in staging, prognostication and targeted molecular therapy. J Urol2005;173(6):1853-1862. [ Links ]
97. Bonsib SM.: The renal sinus is the principal invasive pathway. A prespective study of 100 renal cell carcinomas. Am J Surg Pathol 2004;28:1594-1600. [ Links ]
98. Algaba F, Trias I, Scarpelli M, Boccon-Gibod L, Kirkali Z, Van Poppel H.: Handling and pathology reporting of renal tumor specimens. Eur Urol 2004;45(4):437-443. [ Links ]
99. Algaba F, Alvarez-Argüelles H, Condom E, et al.: Protocolos diagnósticos y pronósticos en Uropatología. Procolos de la Sociedad Española de Anatomía patológica. VB. Madrid, 2001. [ Links ]
100. Van Poppel H, Vandendriessche H, Boel K, Mertens V, Goethuys H, Haustermans K, Van Damme B, Baert L.: Microscopic vascular invasion is the most relevant prognosticator after radical nephrectomy for clinically nonmetastatic renal cell carcinoma. J Urol 1997;158(1):45-49. [ Links ]
101. Merino MJ, Eccles DM, Linehan WM, Algaba F, Zbar B, Kovacs G. Familial renal cell carcinoma. In Eble JN, Sauter G, Epstein JI, Sesterhenn IA. World Health Organization Classification of tumours. Pathology and Genetics Tumours of the urinary system and male genital organs. p-15, IARC Press, Lyon 2004. [ Links ]
102. Maher ER, Kaelin WG.: von Hippel-Lindau disease. Medicine 1997; 76: 381-391. [ Links ]
103. Sampson JR, Patel A, Mee AD.: Multifocal renal cell carcinoma in sibs from a chromosome 9 linked (TSC1) tuberous sclerosis family. J Med Genet. 1995;32(11):848-850. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. F. Algaba
Sección de Patología. Fundació Puigvert
Calle Cartagena 340-350
08025 Barcelona
E-mail: falgaba@fundacio-puigvert.es
Trabajo recibido el 27 de diciembre 2005

