










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActas Urológicas Españolas
versión impresa ISSN 0210-4806
Actas Urol Esp vol.31 no.9 oct. 2007
Tumores renales de la infancia y adolescencia asociados a anomalías cromosómicas
Renal tumors of childhood and adolescence associated with chromosomal anomalies
Cajaiba MM, Reyes-Múgica M.
Program of Pediatric and Developmental Pathology, Yale University School of Medicine. New Haven, USA.
Dirección para correspondencia
RESUMEN
El riñón pediátrico es sitio frecuente de tumores que exhiben alteraciones cromosómicas características. El más común es el nefroblastoma o tumor de Wilms (TW) que se asocia con dos loci: 11p13 (WT1) y 11p15 (WT2 ó BWS), este último ligado también del síndrome de Beckwith-Wiedemann. Otros dos genes que parecen estar implicados son WT3 y WT4; además, dos anomalías específicas (adquisición 1q y deleción 22) se han correlacionado de manera independiente con un peor pronóstico en TW. Otras neoplasias con rearreglos cromosómicos, tales como los carcinomas renales (CRs), son mucho menos frecuentes en niños (entre el 1.8 y el 6.3% de todos los tumores renales malignos). Entre estos, se han identificado "CRs con translocación" que afectan el locus Xp11, siendo los dos tipos más importantes t(X;1), y t(X;17). El nefroma mesoblástico congénito (NMC) es un tumor renal de recién nacidos y lactantes. Los NMCs de la variedad celular se caracterizan por una translocación específica t(12;15)(p13;q25), misma que se encuentra también en los fibrosarcomas congénitos extrarenales, y que permite establecer una correspondencia genética entre estos dos tumores (NMC y fibrosarcoma congénito). Los tumores rabdoides (TR) del riñón son neoplasias muy infrecuentes y muy agresivas, que aparecen con una edad promedio de 11 meses. Al menos 50% de los TRs muestran anormalidades en el gen hSNF5/INI1, situado en el locus 22q11.2. Este gen probablemente está involucrado en la modulación transcripcional de otros genes, tales como el oncogen c-Myc, y de la vía de transducción de la proteína RB-retinoblastoma.
Palabras clave: Citogenética. Carcinoma renal. Tumor de Wilms. Nefroma mesoblástico. Tumor rabdoide renal.
ABSTRACT
The pediatric kidney is a common site for tumors carrying specific chomosomal alterations. The most common of these is the nephroblastoma or Wilms tumor (WT), which is associated with anomalies in two loci: 11p13 and 11p15, the latter also linked to Beckwith-Wiedemann syndrome. Two other genes that seem to be implicated are WT3 and WT4. In addition, 1q gains or 22 deletions have been shown to independently be associated with a worst prognosis in WTs. Other neoplasms with chromosomal rearrangements, such as Renal Cell carcinomas (CRs) are much less frequent in children (between 1.8 and 6.3 % of all malignant renal tumors). Among these, the "translocation renal carcinomas" have been identified involving the locus Xp11 with two main types of translocations: t(X;1), and t(X;17). Congenital mesoblastic nephroma (NMC) is a renal tumor affecting newborns and young infants. NMCs of the cellular type feature a specific translocation t(12;15)(p13;q25), which is also present in congenital fibrosarcomas outside of the kidney. These findings have led to conclude that these two tumors (NMC and congenital fibrosarcoma) are genetically equivalent. Rhabdoid tumors (TRs) of the kidney are very rare and aggressive neoplasms that appear with a mean age of 11 months. At least 50% of these TRs carry abnormalities in the hSNF5/INI1 gene, at 22q11.2. This gene is probably involved in transcriptional modulation of other genes such as the oncogene c-Myc, and also of the retinoblastoma protein RB signaling pathway.
Key words: Cytogenetics. Renal cell carcinoma. Wilms tumor. Congenital mesoblastic nephroma. Rhabdoid tumor of kidney.
Los tumores renales en pacientes adultos son relativamente frecuentes y sus características clínicas y patológicas están bien establecidas; así mismo existen actualmente protocolos terapéuticos que han conseguido una mejor calidad de vida y mayor sobrevivencia. En niños y jóvenes también están bien establecidos los criterios diagnósticos y abordaje terapéutico de los tumores renales, sobretodo del tumor de Wilms; sin embargo, existen otros tumores renales en pacientes pediátricos y adolescentes que, aún siendo menos frecuentes, es necesario revisar, para estar familiarizado con sus alteraciones cromosómicas, publicadas recientemente. Muchas de esas alteraciones deben ser investigadas en el estudio de los tumores renales de la infancia y adolescencia, ya que pueden tener implicaciones en el diagnóstico, pronóstico y tratamiento de esas neoplasias.
En este artículo se revisan fundamentalmente datos histológicos y moleculares de los principales tumores renales de la infancia y adolescencia, incluyéndose el carcinoma de células renales, el tumor de Wilms, el nefroma mesoblástico congénito y el tumor rabdoide renal. Otras neoplasias en las que también se observan alteraciones cromosómicas y que pueden afectar el riñón, tales como las de la familia perteneciente al sarcoma de Ewing/TNEP, los linfomas, y el rabdomiosarcoma, no son incluidas debido a que sólo excepcionalmente tienen localización renal y su estudio pertenece más bien al área de los sarcomas extrarenales, tumores óseos y de tejidos blandos, y al campo de la hematopatología. Hemos excluido también lo referente a tumores tales como el sarcoma de células claras, el angiomiolipoma, o el tumor osificante del riñón infantil, en los que los hallazgos citogenéticas son meramente anecdóticos o no existen.
Carcinomas renales
Los carcinomas renales (CRs) son raros en la niñez y sólo representan entre el 1.8 y el 6.3% de todos los tumores renales malignos. La edad promedio al momento del diagnóstico es de 10 años, y se manifiestan con dolor abdominal, hematuria, o masa palpable, aunque en niños es raro encontrar esta triada clásica. Como en los adultos, el estadio tumoral es el factor pronóstico más importante1-3. En los CRs de niños la morfología de células claras es mucho menos común, mientras que el patrón papilar es más frecuentemente observado que en los casos de adultos. Los carcinomas medulares, aunque raros, ocurren más frecuentemente en niños, casi siempre asociados a enfermedad de células falciformes. Aunque la evolución clínica de los CRs en adultos y en niños es similar, los patrones morfológicos son distintos, y los tumores infantiles no se asocian a defectos genéticos predisponentes tales como síndrome de von Hippel Lindau, como en los adultos. Estas diferencias sugieren que la patogénesis de ambos grupos puede seguir vías oncogenéticas distintas1-4.
Recientemente se ha identificado un grupo de CRs en niños que muestra translocaciones cromosómicas5,6 que involucran un punto de rompimiento específico en Xp11.2. Se ha demostrado que esta región cromosómica alberga el gen TFE3 que codifica un factor de transcripción. Las translocaciones resultan en un gen de fusión en el que TFE3 se combina con promotores de otros genes, lo que probablemente representa un evento de transformación oncogénica en estos tumores, los cuales son algunas veces llamados "carcinomas con translocación Xp11" (Xp11-translocation carcinomas).
Los dos tipos más importantes que caracterizan a este grupo de CRs son t(X;1), y t(X;17). Estas dos translocaciones originan dos transcriptos de fusión diferentes: PRCC-TFE3, y ASPL-TFE3, respectivamente. Otras anomalías cromosómicas que afectan la región Xp11.2 incluyen t(X;1)(p11.2;p34), e inv(X)(p11.2;q12), ambas relacionadas con CRs papilares que muestran fusión de TFE3 con variantes de empalme (splicing variants) de los genes PSF y NonO(p54 nrb)7. Las semejanzas clinicopatológicas (vide infra) que se observan entre los dos grupos mayores de tumores que comparten las mismas alteraciones citogenéticas hacen que estos grupos se sitúen como entidades especiales dentro del espectro de CRs de niños.
Además de las translocaciones Xp11.2, Argani et al.8 han descrito la translocación t(6;11) (p21;q12) en otros CRs con fusión de los genes α (11q12) y TFEB (6p21). El fenotipo de tumores con esta alteración tiene características típicas (vide infra). Esta anormalidad provoca la expresión anómala de TFEB9. Es interesante mencionar que tanto TFE3 como TFEB pertenecen a la misma familia de factores de transcripción.
Las alteraciones citogenéticas relacionadas a CRs pueden ser detectadas por medio de cariotipo convencional, FISH y RT-PCR. A través de inmunohistoquímica puede detectarse la expresión nuclear tanto de TFE3 como de TFEB en tumores que tienen translocaciones Xp11.2 y 6;11, respectivamente, permitiendo estudios utilizando tejido incluido en parafina (Argani 2003 y 2005)8,10.
CRs con fusión ASPL-TFE3
Este transcripto resulta de la translocación t(X;17)(p11.2;q25)5,11. Es interesante que los sarcomas alveolares de partes blandas (ASPSs, por su siglas en inglés), muestran de manera característica una anomalía citogenética similar, involucrando los mismos puntos de rompimiento cromosómico, aunque de manera distinta a los CRs, pues en éstos últimos la translocación es balanceada, mientras que en aquellos es no balanceada, generando una dosis genética diferente12. Aunque este tipo de CRs comparte características genéticas y morfológicas con los ASPSs (vide infra), estas dos neoplasias difieren en sus rasgos ultraestructurales y perfil de expresión genética, por lo que no pueden considerarse la misma entidad.
La edad de presentación de pacientes con este tumor renal oscila entre 17 meses y 17 años, y la mayoría de los casos se presentan en estadios clínicos avanzados [NWTS-5 (National Wilms Tumor Study-5), estadios 3 y 4]. Sus características microscópicas (algunas de las cuales comparten con los ASPSs) incluyen un patrón organoide, con arquitectura de láminas celulares compuestas de nidos celulares sólidos, y delicados septos fibrovasculares; también puede observarse un patrón seudopapilar con calcificaciones psamomatosas (Figs. 1 y 2). Usualmente hay necrosis y hemorragia multifocales, y la invasión linfática y vascular, así como las metástasis a ganglios linfáticos son frecuentes. Las células pueden aparecer focalmente sin cohesión, con un citoplasma predominantemente claro o finamente granular, y puede haber atipia nuclear moderada con nucléolos prominentes5,11. El inmunofenotipo incluye expresión focal para antígeno de membrana epitelial (EMA), citoqueratinas y S-100. La microscopía electrónica muestra células con gránulos electrodensos y cristales similares a los observados en ASPSs, así como rasgos de diferenciación epitelial, tales como uniones intercelulares, membrana basal y luces glandulares5.
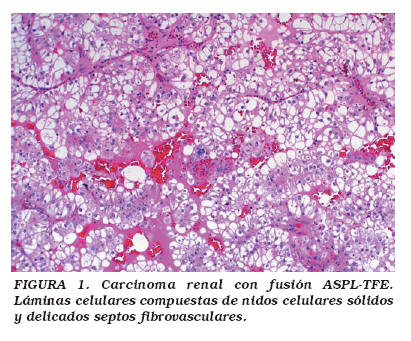
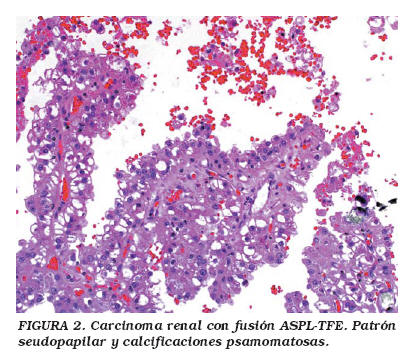
CRs con fusión PRCC-TFE3
Estos CRs presentan translocación t(X;1) (p11.2;q21), originando fusión entre TFE3 y PRCC, localizado, éste último gen, en el locus 1q21. Tumores de este tipo han sido observados tanto en adultos como en niños, pero casi la mitad de ellos han aparecido entre los 2 y los 15 años de edad6,13-15. La mayoría de los casos han sido clasificados en el NWTS-5 en estadios 1 ó 26.
Histológicamente, todos los tumores con fu-sión PRCC-TFE3 muestran algún grado de diferenciación papilar entremezclado con áreas sólidas que exhiben nidos, acinos y/o túbulos; usualmente las áreas papilar y sólida se encuentran claramente demarcadas. Las células en ambas áreas pueden mostrar citoplasma granular o eosinofílico, y la mayoría de los tumores tienen un bajo grado de atipia nuclear (equivalente al grado 2 de Fuhrman) con núcleos redondeados y sin nucléolos. Hay pocas mitosis y la invasión vascular es poco frecuente. Tanto la necrosis, como la hemorragia y los cuerpos psamomatosos son menos frecuentes que en los tumores con fusión de ASPL-TFE36,13,15.
El inmunofenotipo incluye expresión de TFE3, RCC y CD10; la citoqueratina, EMA y vimentina son negativas o sólo focalmente positivas en la mayoría de los casos. S-100, HMB45 y desmina son siempre negativas. Ultraestructuralmente hay rasgos de CRs convencionales, tales como uniones intercelulares, mitocondrias abundantes, y glucógeno; algunos tumores han mostrado microtúbulos dentro de cisternas, parecidos a los descritos en melanomas6.
CRs con fusión α-TFEB
Los tumores con esta fusión resultan de la translocación t(6;11)(p21;q12), la cuál se ha informado también en adultos (la edad de los pacientes oscila entre 9 y 33 años), con edad media al diagnóstico de 17 años8. No hay datos definitivos respecto a su estadio clínico o pronóstico.
Los tumores α-TFEB muestran un fenotipo notable, caracterizado por una población celular bifásica y la presencia de numerosos nódulos hialinos. Estos tumores exhiben una arquitectura sólida o en nidos, con pocas estructuras tubulares y papilares. El principal tipo celular es epitelioide, con citoplasma eosinofílico abundante, y atipia nuclear consistente con un grado 3 de Fuhrman. El segundo tipo celular, menos numeroso, se caracteriza por células pequeñas de cromatina densa, usualmente agrupadas alrededor de los nódulos hialinos. Puede observarse invasión vascular. Inmunohistoquímicamente, estas neoplasias también son interesantes. Las células no expresan antígenos epiteliales tales como EMA o queratinas, pero los marcadores melanocíticos como HMB45 y Melan-A son consistentemente positivos. Otros marcadores tales como RCC y S-100 son negativos. La microscopía electrónica muestra abundante membrana basal y otras características epiteliales, incluyendo complejos de unión intercelular, luces glandulares y microvellosidades.
Tumor de Wilms (nefroblastoma)
Las neoplasias renales de los niños representan un conjunto heterogéneo, tanto clínica como fenotípicamente16,17. Entre éstas, el tumor de Wilms (TW) o nefroblastoma es la más común y uno de los tumores más frecuentes en la niñez, representándo el 6.3% del total de acuerdo al programa de vigilancia epidemiológica del NCI- [Surveillance, Epidemiology and End Results (SEER) program]. A principios del siglo XX, la mortalidad de niños afectados por cáncer renal sobrepasaba el 90%; un siglo después, en los albores del siglo XXI, la sobrevida a cinco años alcanza alrededor del 90%.
En gran medida, este éxito depende de una adecuada evaluación macro y microscópica por parte del patólogo. Recientemente, Bruce Beckwith, uno de los responsables de los grandes avances en relación al nefroblastoma, y por muchos años el patólogo al frente del Grupo Nacional de Estudio del Tumor de Wilms en los EEUU (NWTSG), se refirió al hecho de que cada vez con mayor frecuencia son los patólogos generales los que confrontan estas lesiones en el laboratorio de patología quirúrgica, a diferencia de lo que ocurría hace algunos años cuando la mayoría de estos casos llegaban a manos de patólogos pediatras en centros académicos (comunicación verbal). Por lo tanto, es fundamental que el patólogo general se familiarice con el protocolo adecuado para el estudio de los tumores de Wilms.
El TW ocurre más frecuentemente entre los 2 y 6 años de vida, y en su mayoría estos casos son detectados por los padres o los pediatras en exploraciones rutinarias. Ocasionalmente puede presentarse con hematuria, hipertensión, o hemorragia aguda después de trauma abdominal, seguida de dolor abdominal súbito. En algunos pacientes, hay historia clínica de síndromes que predisponen a neoplasias, tales como el síndrome de Bloom18-20. Aunque en algunos casos, y especialmente en Europa, se opta por la quimioterapia preoperatoria, la mayor parte de estos tumores, por lo menos en el continente americano son resecados con nefrectomía, la cual debe permitir una adecuada estadificación, así como información sobre histología favorable o desfavorable, lo cual impacta notablemente el tratamiento y pronóstico.
Típicamente el TW es una masa grande, abultada y que distorsiona el contorno renal (Fig. 3). El espécimen debe pesarse antes de proceder a la disección. Al corte, la masa hace prominencia sobre la superficie, y es muy importante teñir con tinta china la cápsula renal antes de cortar, pues la cápsula se retrae con el corte, modificando así la relación de esta estructura con el tumor. El corte debe iniciarse desde el hilio renal para evitar la contaminación artificial del seno renal con fragmentos de tumor arrastrados por el cuchillo. La superficie de corte muestra áreas alternantes de hemorragia, necrosis y tumor carnoso. Deben tomarse cortes representativos del tumor, así como del riñón no tumoral con objeto de detectar restos nefrogénicos. Es de particular importancia evaluar la interfase tumor-parénquima en la región del seno renal. Esta zona es crítica para la adecuada estadificación, ya que ahí no existe la cápsula renal y además es la zona de mayor vascularizacion eferente por la cual el tumor inicia su trayecto metastásico fuera del riñón16.
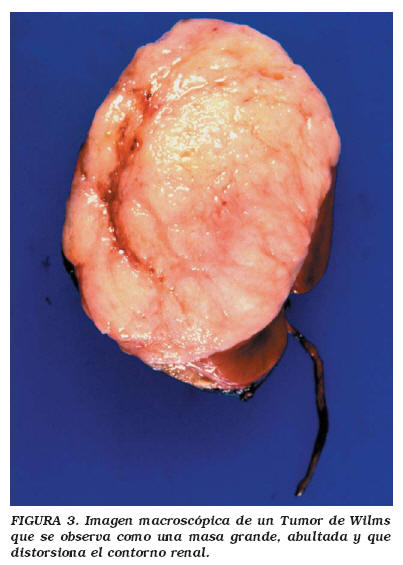
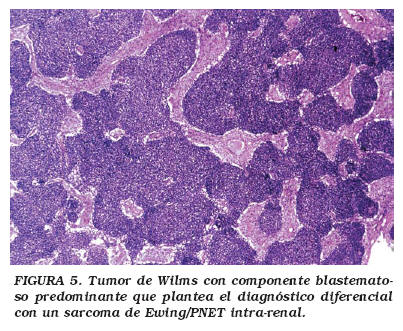
Microscópicamente, el TW muestra grados variables de la combinación de tres elementos: blastema, epitelio y estroma (Fig. 4). El blastema es el menos diferenciado de los tres, formado por células "pequeñas redondas y azules", y del cual se derivan los otros dos componentes. Cuando el blastema es el componente predominante, debe establecerse el diagnóstico diferencial con un sarcoma de Ewing/PNET intra-renal (Fig. 5). El componente epitelial forma túbulos primitivos. Con frecuencia pueden observarse glomérulos primitivos. El estroma muestra células fusiformes mesenquimatosas. Cualquiera de los tres componentes puede predominar sobre los otros, y ocasionalmente ser el componente exclusivo. Pueden también encontrarse componentes heterólogos tales como músculo liso o estriado, tejido adiposo, cartílago, hueso o tejido neuroglial, produciendo ocasionalmente un patrón referido como TW teratoide. Cuando el TW contiene un componente abundante (por lo menos del 30%) de músculo estriado, se le denomina nefroblastoma rabdomiomatoso fetal, el cual es más frecuentemente bilateral y de curso clínico relativamente benigno. Otra variante del TW es la lesión quística. Ocasionalmente el TW convencional puede mostrar múltiples quistes, sin embargo, cuando estos son predominantes y exhiben en sus paredes pequeños focos de blastema, la lesión se designa como nefroblastoma quístico parcialmente diferenciado, cuyo curso clínico es también relativamente benigno, con estadios clínicos tempranos. El extremo benigno de este espectro está representado por el nefroma quístico, compuesto por quistes de tamaño variable pero sin elementos blastematosos21.
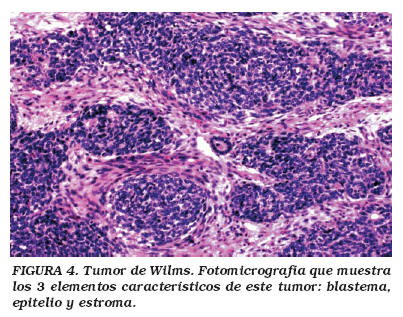
Immunohistoquímica
Aunque existen algunos anticuerpos que reaccionan con las células del TW, la utilidad práctica de la inmunohistoquímica en el diagnóstico de esta entidad es muy limitada. La mayoría de los nefroblastomas expresan citoqueratina, CD56 y desmina, y una pequeña proporción son positivos para CD99 y actina muscular-específica. Anti-cuerpos de generación más reciente, tales como el dirigido contra la proteína WT1 están empezando a utilizarse21.
Microscopía electrónica
Los rasgos ultraestructurales del TW incluyen formación de luces, microvellosidades, cilios, y en particular, una capa densa de lámina basal en la superficie luminal. Estos hallazgos pueden apoyar el diagnóstico, pero no son totalmente confiables en caso de biopsias pequeñas y con tumores poco diferenciados21.
Histología pronóstica
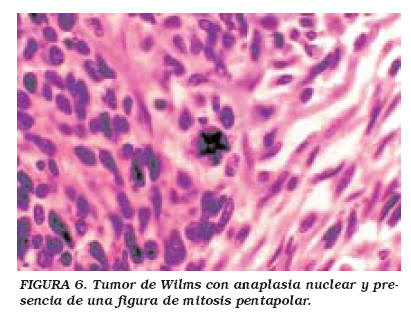
Uno de los signos microscópicos mas importantes de identificar en los tumores de Wilms es la presencia de anaplasia (Fig. 6), definida como la presencia de figuras mitósicas anormales (aneuploides) y/o núcleos hipercromáticos y de un diámetro por lo menos tres veces mayor que el de núcleos de células adyacentes22. La anaplasia se asocia a mal pronóstico debido a que estas células son más resistentes a la quimioterapia. Sin embargo, los tumores de Wilms anaplásicos no son más agresivos, ni muestran mayor capacidad metastásica o de invasión tisular. La anaplasia debe clasificarse como focal o difusa. Sólo la anaplasia difusa se asocia con mal pronóstico, ya que por definición, la anaplasia focal se observa solamente en el tumor resecado, y las células resistentes son extirpadas como parte del tumor16. La única excepción es el TW con anaplasia focal en estadio I, que recibe un tratamiento combinado de cirugía y quimioterapia, a diferencia de la variante no anaplásica, que sólo amerita extirpación quirúrgica. La anaplasia puede ser detectada o confirmada analizando la ploidia tumoral mediante citometría de flujo23.
Otro hallazgo que es importante identificar es la presencia de restos nefrogénicos, comúnmente llamados "nefroblastomatosis" (aunque esta designación es incorrecta)24. Estos focos se encuentran en áreas de riñón no tumoral en más del 30% de los casos, y pueden ser perilobares y/o intralobares; más raramente puede haber afección panlobar. Su presencia se asocia con la posibilidad de un TW en el riñón contralateral, por lo cual es muy importante hacer un muestreo adecuado. El tipo perilobar se asocia con tumor contralateral sincrónico de tipo blastematoso y epitelial; la variante intralobar se asocia con tumor contralateral metacrónico y de predominio estromal. Estos restos nefrogénicos representan lesiones dinámicas, y pueden ser hiperplásicos, neoplásicos, obsolescentes o escleróticos16.
Biología molecular
La asociación del TW con diversas anomalías congénitas tales como anirida, hemihipertrofia y malformaciones genitourinarias, conformando síndromes con diversos patrones de herencia ha servido de estímulo para lograr avances en el entendimiento de la biología molecular de esta neoplasia25-27. De este modo, se han localizado dos loci cromosómicos asociados a TW: 11.13 (WT1) y 11.15 (WT2 ó BWS), este último responsable también del síndrome de Beckwith-Wiedemann. Otros dos genes parecen estar implicados en por los menos algunos casos de TW (WT3 y WT4); sin embargo, en el terreno del diagnóstico anatomopatológico, la aplicación de técnicas de biología molecular aún no adquiere una utilidad práctica25-33.
Entre el 60 y el 82% de TW estudiados muestra cariotipos anormales34-37. Las anomalías citogenéticas en el nefroblastoma con frecuencia son complejas, caracterizadas por la aparición concomitante de distintas alteraciones. El estudio de pérdida de heterocigosidad (LOH) ha sido útil para encontrar pérdidas cromosómicas38,39.
Las anomalías genéticas más frecuentes se mencionan a continuación34-37:
1. Anormalidades numéricas (que afectan ploidía):
- Polisomías (trisomías) 6, 8, 12 y 18.
- Deleción de cromosoma 22 (monosomía).
2. Anormalidades estructurales:
- Deleción de 1p, puede resultar de isocromosoma 1q, translocación no balanceada y cromosoma 1 en anillo.
- Deleción de 11p, que resulta de pérdidas parciales o totales del cromosoma 11.
- Deleción 16q, que resulta de deleciones, o translocaciones con el cromosoma 1 principalmente.
- Adición 1q, que resulta de translocaciones no balanceadas, especialmente con 16q, y por isocromosoma 1q.
Se ha observado que los niños con anormalidades genéticas son significativamente mayores que aquellos que tienen cariotipos normales, lo cual sugiere diferencias biológicas entre los dos grupos35,36. Sin embargo, el análisis de dos grandes series (tumores con cariotipos normales y otros con cariotipos anormales) no demostró diferencias estadísticamente significativas en características histológicas, estadio tumoral o evolución clínica35-37. En una serie grande de Brown et al.36 dos anomalías específicas (adquisición 1q y deleción 22) correlacionaron de manera independiente con un peor pronóstico.
Recientemente, algunos estudios han demostrado una asociación entre LOH y la evolución clínica de TW. Yuan et al (2005)38 mostró que estadios clínicos avanzados correlacionaron con incremento en el número de regiones con LOH, reflejando anormalidades citogenéticas más complejas. Así mismo, TW de histología favorable con LOH para ambas regiones, 1p y 16q, mostraron tener un incremento relativo en el riesgo de recurrencias en un estudio del NWTS, y estos hallazgos se usan actualmente para la estratificación de pacientes y su asignación a protocolos terapéuticos40.
En resumen, las anormalidades citogenéticas son un hallazgo común en el TW, y probablemente desempeñan un papel importante en sus características biológicas. Sin embargo, no es posible establecer correlaciones firmes entre hallazgos moleculares o citogenéticos y características clínicas, debido a resultados limitados y hasta conflictivos ofrecidos por diferentes grupos. Hoy por hoy, el papel de la citogenética en el abordaje terapéutico del TW es poco claro, y aunque se han hecho intentos por establecer un valor pronóstico para anomalías genéticas específicas, el cariotipo de rutina no agrega información de utilidad práctica al informe del patólogo.
Nefroma mesoblástico congénito
El nefroma mesoblástico congénito (NMC) típicamente aparece en el riñón de recién nacidos y lactantes de hasta 1 año de edad41-44. La edad promedio al momento del diagnóstico es de 2 meses. Los pacientes usualmente se presentan con una masa abdominal palpable. Otras manifestaciones incluyen polihidramnios durante el embarazo, hipertensión y hematuria. El tratamiento es quirúrgico, ya que el NMC tiene un buen pronóstico a pesar de que se ha informado la existencia de recurrencias locales y hasta de metástasis.
Patología
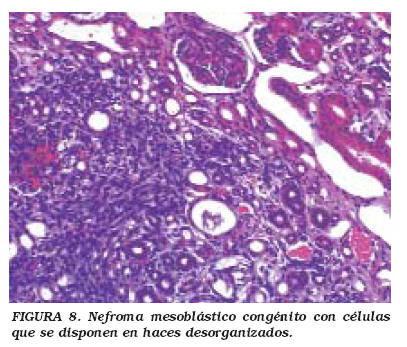
La mayoría de los NMC aparecen en la región central cercana al hilio renal. Microscópicamente son tumores bien delimitados, homogéneos y firmes. Pueden clasificarse en dos variedades: la clásica y la celular (atípico); hay ejemplos donde ambos tipos coexisten. El NMC clásico es un tumor monoformo, fusocelular, con haces anastomosantes de células con aspecto de fibroblasto o miofibroblasto, y con fibras colágenas. La proliferación neoplásica "atrapa" tejido renal normal en el que con frecuencia se observan cambios metaplásicos y displásicos. El tumor se extiende con bordes que "empujan", sin una delimitación o interfase muy clara entre el tumor y el parénquima renal. La atipia celular, las mitosis o la necrosis son excepcionales. En contraste, la variedad celular (o atípica) frecuentemente exhibe zonas macroscópicas de necrosis, hemorragia, reblandecimiento y degeneración quística. Histológicamente (Figs. 7 y 8), el NMC celular muestra mayor densidad celular, pleomorfismo y aumento de la relación núcleo/citoplasma. Las células se disponen en haces desorganizados, y la actividad mitósica usualmente supera 8-10 mitosis/campo de alto aumento43,44.
Inmunohistoquímica y microscopía electrónica
El inmunofenotipo es inespecífico, con expresión de marcadores miofibroblásticos en la mayoría de los casos, incluyendo vimentina, actina y desmina. La ultraestructura es la de una lesión miofibroblástica, lo cual puede ser de utilidad en el diagnóstico43,44.
Citogenética/Biología Molecular
Los NMCs celulares se caracterizan por una transolación específica t(12;15)(p13;q25), que se encuentra virtualmente en todos los casos. En contraste con lo anterior, los NMCs clásicos puros no exhiben esta translocación cromosómica45,48. Debe mencionarse que esta cromosomopatía, así como otras características clínicas y fenotípicas son idénticas a las observadas en otro tumor de la infancia temprana: el fibrosarcoma congénito (FSC). Por lo tanto, la tendencia actual es a considerar que estas dos neoplasias, NMC y FSC son la variante renal y de tejidos blandos, respectivamente, de la misma entidad45,49.
La t(12;15)(p13;q25) resulta en la fusión del gen ETV6 (situado en el locus 12p13), el cual codifica un factor de transcripción, con el gen NTRK3 (locus 15q25), que codifica un receptor de membrana de tipo quinasa de tirosina. El potencial oncogénico adquirido por este trascriptosoma de fusión parece estar involucrado en la patogénesis de los NMCs y los FSCs, y puede relacionarse con la progresión de NMCs tipo clásico a tipo celular. Otras alteraciones citogenéticas tales como trisomías 8, 11, 17 y 20, ocurren menos frecuentemente, tanto en NMCs celulares como en FSCs45,49,50.
Estudios enfocados a detectar la presencia de la t(12;15)(p13;q25) en casos de NMCs celulares tienen utilidad sólo diagnóstica, ya que esta anormalidad no predice diferencias en comportamiento clínico48. Los estudios citogenéticos en NMCs resultan de gran utilidad para establecer el diagnóstico diferencial con otros tumores del riñón en niños muy pequeños, especialmente tumor de Wilms de predominio blastematoso, así como sarcoma de células claras, pues estas neoplasias no muestran la translocación. Debe mencionarse que estudios citogenéticos de alta resolución deben ser utilizados cuando se intenta demostrar la t(12;15)(p13;q25), ya que su patrón de bandeo es sutil y puede pasar inadvertida si sólo se usan métodos citogenéticos convencionales (cariotipo)45,48,51.
Tumor Rabdoide
Los tumores rabdoides (TR) del riñón son neoplasias raras y clínicamente muy agresivas. La mayoría se presenta en el primer año de vida, aunque puede verse desde la etapa neonatal hasta los 8 años de edad, con una edad promedio de 11 meses. Estos tumores comparten similitudes clínicas, fenotípicas y biológicas con tumores rabdoides que afectan otros sitios anatómicos, incluyendo los del Sistema Nervioso Central (SNC). Generalmente, al momento del diagnóstico, el estadio clínico es avanzado, y la tasa general de sobrevida es baja. Tanto el estadio clínico avanzado como una edad de presentación muy temprana son considerados factores de mal pronóstico. Usualmente se manifiestan con una masa abdominal palpable, frecuentemente acompañada de hipercalcemia. Algunos pacientes pueden también presentarse con un tumor primario del SNC, usualmente meduloblastomas, PNET o astrocitomas. Los pulmones son el sitio más común de metástasis52-54.
Patología
Macroscópicamente, los TRs son tumores grandes que reemplazan el riñón, y muestran una superficie homogénea al corte, con focos de necrosis y hemorragia; es común encontrar nódulos satélites. Histológicamente, la arquitectura es sólida compuesta de láminas de células sin cohesión, con abundante citoplasma eosinófilo que desplaza un núcleo vesicular con nucléolo prominente; pueden observarse mitosis numerosas. Una característica clásica aunque variable es la presencia de inclusiones paranucleares eosinofílicas, hialinas y globulares. Frecuente-mente se observa invasión vascular, extensión hacia el seno renal, cápsula renal y parénquima renal adyacente, atrapando glomérulos y túbulos que ocasionalmente exhiben cambios metaplásicos como los observados en los Nefromas Mesoblásticos Congénitos. Algunos TRs pueden tener un patrón esclerosante con nidos de células separadas por un estroma hialino semejante a osteoide52,53.
Inmunohistoquímica y microscopía electrónica
El inmunofenotipo es inespecífico, con expresión de vimentina, y menos frecuentemente de algunos marcadores epiteliales tales como citoqueratinas y antígeno de membrana epitelial (EMA). Los marcadores de diferenciación muscular son negativos. La miscroscopía electrónica muestra inclusiones citoplásmicas características compuestas por agregados de filamentos intermedios dispuestos en un patrón compacto y arremolinado; otras hallazgos ultrastructurales son los nucléolos prominentes e uniones intercelulares rudimentarias52,53,55.
Citogenética y Biología molecular
Al menos 50% de los TRs muestran anormalidades en el gen hSNF5/INI1, situado en el locus 22q11.2.56,57 Este gen también está inactivado en la mayoría de los TRs extrarenales, y estas alteraciones genéticas parecen ser específicas de esta familia de tumores. El hSNF5/INI1 es un gen que participa en la remodelación de cromatina, y probablemente está involucrado en la modulación transcripcional de otros genes tales como el oncogen c-Myc, y de la vía de transducción de la proteína RB (retinoblastoma)58.
Las anormalidades de hSNF5/INI1 en los TRs, al igual que lo que ocurre con otros genes supresores de tumores, afectan los dos alelos. Se han descrito deleciones, mutaciones y pérdida de la heterocigocidad (LOH). Asímismo, un porcentaje significativo de TRs demuestra mutaciones de línea germinal caracterizando un síndrome de predisposición a cáncer en el cual los niños tienen un riesgo aumentado de desarrollar otros TRs primarios en diferentes sitios anatómicos59; en estos casos, se han encontrado mutaciones de novo más que herencia de alelos mutados56,57.
El hallazgo de anormalidades moleculares de hSNF5/INI1 en TRs tiene un papel importante principalmente en la confirmación del diagnóstico, permitiendo eliminar otras posibilidades en el diferencial con otros tumores. Sin embargo, la confirmación de inactivación de hSNF5/INI1 no proporciona información sobre el pronóstico u opciones terapéuticas en casos de TRs, aunque se ha discutido la posibilidad de usar agentes remodeladores de cromatina en el tratamiento de TRs58,60.
Las alteraciones más frecuentemente informadas en hSNF5/INI1 son mutaciones puntuales, y éstas pueden detectarse por medio de secuenciación directa. Deleciones que producen inactivación alélica pueden detectarse por hibridación in situ fluorescente (FISH) o PCR56,57. Los estudios de inmunohistoquímica en parafina usando anticuerpos contra hSNF5/INI1 son muy útiles en el diagnóstico de TRs, pues la expresión de este gen es consistentemente negativa en estos casos, mientras que la mayoría de los otros tumores renales son positivos61,62. La ausencia de expresión de hSNF5/INI1 por inmunohistoquímica aún en casos sin deleciones o mutaciones en el gen sugiere que otros mecanismos además de los anteriormente citados, pueden ser responsables de su inactivación61.
Referencias
1. Indolfi P, Terenziani M, Casale F, Carli M, Bisogno G, Schiavetti A, et al. Renal cell carcinoma in children: a clinicopathologic study. J Clin Oncol. 2003;21(3):530-535. [ Links ]
2. Murphy WM, Grignon DJ, Perlman EJ. Kidney tumors in children .Pediatric renal cell carcinoma. In: Tumors of the Kidney, Bladder, and Related Urinary Structures. Washington, DC: Armed Forces Institute of Pathology;2004:82-88. [ Links ]
3. Selle B, Furtwangler R, Graf N, Kaatsch P, Bruder E, Leuschner I. Population-based study of renal cell carcinoma in children in Germany, 1980-2005: more frequently localized tumors and underlying disorders compared with adult counterparts. Cancer. 2006;107(12):2906-2914. [ Links ]
4. Bruder E, Passera O, Harms D, Leuschner I, Ladanyi M, Argani P, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol. 2004; 28(9):1117-1132. [ Links ]
5. Argani P, Antonescu CR, Illei PB, Lui MY, Timmons CF, Newbury R, et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol. 2001; 159(1):179-192. [ Links ]
6. Argani P, Antonescu CR, Couturier J, Fournet JC, Sciot R, Debiec-Rychter M, et al. PRCC-TFE3 renal carcinomas: morphologic, immunohistochemical, ultrastructural, and molecular analysis of an entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol. 2002;26(12):1553-1566. [ Links ]
7. Clark J, Lu YJ, Sidhar SK, Parker C, Gill S, Smedley D, et al. Fusion of splicing factor genes PSF and NonO(p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene. 1997;15(18):2233-2239. [ Links ]
8. Argani P, Lae M, Hutchinson B, Reuter VE, Collins MH, Perentesis J, et al. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol. 2005;29(2):230-240. [ Links ]
9. Kuiper RP, Schepens M, Thijssen J, van Asseldonk M, van den Berg E, Bridge J,et al. Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum Mol Genet. 2003;12(14):1661-1669. [ Links ]
10. Argani P, Lal P Hutchinson B, Lui MY, Reuter VE, Ladanyi M. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol. 2003;27(6): 750-761. [ Links ]
11. Zambrano E, Reyes-Múgica M. Renal cell carcinoma with t(X;17): singular pediatric neoplasm with specific phenotype/genotype features. Ped Dev Pathol. 2002;6(1):84-87. [ Links ]
12. Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20(1):48-57. [ Links ]
13. De Jong B, Molenaar IM, Leeuw JA, Idenberg VJS, Oosterhuis JW. Cytogenetics of a renal adenocarcinoma in a 2-year-old child. Cancer Genet Cytogenet. 1986;21(2):165-169. [ Links ]
14. Meloni AM, Dobbs RM, Pontes JE, Sandberg AA. Translocation (X;1) in papillary renal cell carcinoma. A new cytogenetic subtype. Cancer Genet Cytogenet 1993;65(1):1-6. [ Links ]
15. Dijkhuizen T, van den Berg E, Storkel S, Terpe HJ, Burger H, de Jong B. Distinct features for chromophilic renal cell cancer with 11.2 breakpoints. Cancer Genet Cytogenet. 1998;104(1):74-76. [ Links ]
16. Beckwith JB. National Wilms Tumor Study: an update for pathologists. Pediatr Dev Pathol 1998;1(1):79-84. [ Links ]
17. Cosentino C, Raffensperger J, Luck S, Reynolds M, Sherman J, Reyes-Múgica M. A 25- year experience with renal tumors of childhood. Journal of Pediatric Surgery 1993;28(10):1350-1355. [ Links ]
18. Cairney AE, Andrews M, Greenberg M, Smith D, Weksberg R. Wilms tumor in three patients with Bloom syndrome. J Pediatr. 1987;111(3):414-416. [ Links ]
19. Berger C, Frappaz D, Leroux D, Blez F, Vercherat M, Bouffet E, et al. Wilms tumor and Bloom syndrome. Arch Pediatr 1996;3(8):802-805. [ Links ]
20. Jain D, Hui P, McNamara J, Schwartz D, German J, Reyes-Múgica M. Bloom syndrome in sibs: first reports of hepatocellular carcinoma and Wilms tumor with documented anaplasia and nephrogenic rests. Pediatr Dev Pathol. 2001;4(6):585-589. [ Links ]
21. Parham DM. Renal Neoplasms. In: Parham DM, ed. Pediatric Neoplasia. Morphology and Biology. Philadelphia: Lippincott-Raven Publishers, 1996:33-64. [ Links ]
22. Bonadio JF, Storer B, Norkool P, Farewell VT, Beckwith JB, D'Angio GJ. Anaplastic Wilms tumor: clinical and pathologic studies. J Clin Oncol. 1985; 3(4):513-520. [ Links ]
23. Douglass EC, Look AT, Webber B, Parham D, Wilimas JA, Green AA, et al. Hyperdiploidy and chromosomal rearrangements define the anaplastic variant of Wilms' tumor. J Clin Oncol 1986; 4:975-81. [ Links ]
24. Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr Pathol. 1990;10(1-2):1-36. [ Links ]
25. Knudson AG Jr. Introduction to the genetics of primary renal tumors in children. Med Pediatr Oncol. 1993; 21(3):193-198. [ Links ]
26. Feinberg AP. Multiple genetic abnormalities of 11p15 in Wilms' tumor. Med Pediatr Oncol. 1996;27(5):484-489. [ Links ]
27. Thorner PS, Squire JA. Molecular genetics in the diagnosis and prognosis of solid pediatric tumors. Pediatr Dev Pathol. 1998;1(5):337-365. [ Links ]
28. Besnard.Guerin C, Newsham I, Winqvist R, Cavenee WK. A common region of loss of heterozygosity in Wilms' tumor and embryonal rhabdomyosarcoma distal to the D11S988 locus on chromosome 11p15.5. Hum Genet. 1996;97(2):163-170. [ Links ]
29. Brown KW, Villar AJ, Bickmore W, Clayton-Smith J, Catchpoole D, Maher ER, et al. Imprinting Mutation in the Beckwith-Wiedemann Syndrome Leads to biallelic Igf2 expression through an H19-independent pathway. Human Molecular Genetics. 1996;5(12):2027-2032. [ Links ]
30. Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, et al. An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet. 1996;14(2):171-173. [ Links ]
31. Mannens M, Alders M, Redeker B, Bliek J, Steenman M, Wiesmeyer C, et al. Positional cloning of genes involved in the Beckwith-Wiedemann syndrome, hemihypertrophy, and associated childhood tumors. Med Pediatr Oncol. 1996;27(5):490-494. [ Links ]
32. Overall ML, Spencer J, Bakker M, Dziadek M, Smith PJ. p57k1p2 is expressed in Wilms' tumor with Loh of 11p15.5. Genes Chromosomes Cancer. 1996;17(1):56-59. [ Links ]
33. Weksberg R, Squire JA. Molecular biology of Beckwith-Wiedemann syndrome. Med Pediatr Oncol. 1996;27(5):462-469. [ Links ]
34. Slater RM, Mannens MM. Cytogenetics and molecular genetics of Wilms' tumor of childhood. Cancer Genet Cytogenet. 1992;61(2):111-121. [ Links ]
35. Gow KW, Murphy JJ. Cytogenetic and histologic findings in Wilms' tumor. J Pediatr Surg. 2002;37(6):823-827. [ Links ]
36. Bown N, Cotterill SJ, Roberts P, Griffiths M, Larkins S, Hibbert S, et al. Cytogenetic abnormalities and clinical outcome in Wilms tumor: a study by the U.K. cancer cytogenetics group and the U.K. Children's Cancer Study Group. Med Pediatr Oncol. 2002;38(1):11-21. [ Links ]
37. Kullendorf CM, Soller M, Wiebe T, Mertens F. Cytogenetic findings and clinical course in a consecutive series of Wilms tumors. Cancer Genet Cytogenet. 2003;140(1): 82-87. [ Links ]
38. Yuan E, Li CM, Yamashiro DJ, Kandel J, Thaker H, Murty VV, et al. Genomic profiling maps loss of heterozygosity and defines the timing and stage dependence of epigenetic and genetic events in Wilms' tumors. Mol Cancer Res. 2005;3(9):493-502. [ Links ]
39. Ruteshouser EC, Hendrickson BW, Colella S, Krahe R, Pinto L, Huff V. Genome-wide loss of heterozygosity analysis of WT1-wild type and WT1-mutant Wilms tumors. Genes Chromosomes & Cancer. 2005;43(2):172-180. [ Links ]
40. Grundy PE, Breslow NE, Li S, Perlman E, Beckwith JB, Ritchey ML, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2005;23(29): 7312-7321. [ Links ]
41. Bolande RP, Brough AJ, Izant RJ Jr. Congenital mesoblastic nephroma of infancy. A report of eight cases and the relationship to Wilms' tumor. Pediatrics. 1967;40(2):272-278. [ Links ]
42. Bolande RP. Congenital mesoblastic nephroma of infancy. Perspect Pediatr Pathol. 1973;1:227-250. [ Links ]
43. Pettinato G, Manivel JC, Wick MR, Dehner LP. Classical and cellular (atypical) congenital mesoblastic nephroma: a clinicopathologic, ultrastructural, immunohistochemical, and flow cytometry study. Hum Pathol. 1989;20(7):682-690. [ Links ]
44. Murphy WM, Grignon DJ, Perlman EJ. Kidney tumors in children. Congenital mesoblastic nephroma. In: Tumors of the Kidney, Bladder, and Related Urinary Structures. Washington, DC: Armed Forces Institute of Pathology; 2004;57-65. [ Links ]
45. Rubin BP, Chen CJ, Morgan TW, Xiao S, Grier HE, Kozakewich HP, et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol. 1998;153(5):1451-1458. [ Links ]
46. Kzenevich SR, Garnett MJ, Pysher TJ, Beckwith JB, Grundy PE, Sorensen PH. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998;58(22):5046-5048. [ Links ]
47. Watanabe N, Kobayashi H, Hirama T, Kikuta A, Koizumi S, Tsuru T, et al. Cryptic t(12;15)(p13;q26) producing the ETV6-NTRK3 fusion gene and no loss of IGF2 imprinting in congenital mesoblastic nephroma with trisomy 11: fluorescence in situ hybridization and IGF2 allelic expression analysis. Cancer Genet Cytogenet. 2002;136(1):10-16. [ Links ]
48. Anderson J, Gibson S, Sebire NJ. Expression of ETV6-NTRK3 in classical, cellular and mixed subtypes of congenital mesoblastic nephroma. Histopathology. 2006;48(6):748-753. [ Links ]
49. Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: congenital (infantile) fibrosarcoma and mesoblastic nephroma. Cancer Genet Cytogenet 2002;132(1):1-13. [ Links ]
50. Speleman F, van den Berg E, Dhooge C, Oosterhuis W, Redeker B, De Potter CR, et al. Cytogenetic and molecular analysis of cellular atypical mesoblastic nephroma. Genes Chromos Cancer. 1998;21(3):265-269. [ Links ]
51. Argani P, Fritsch M, Kadkol SS, Schuster A, Beckwith JB, Perlman EJ. Detection of the ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: application to challenging pediatric renal stromal tumors. Mod Pathol. 2000;13(1):29-36. [ Links ]
52. Weeks D, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney: a report of 111 cases from the National Wilms Tumor Study Pathology Center. Am J Surg Pathol. 1989;13(6):439-458. [ Links ]
53. Vujanic GM, Sandstedt B, Harms D, Boccon-Gibod L, Delemarre JFM. Rhabdoid tumour of the kidney: a clinicopathological study of 22 patients from the International Society of Pediatric Oncology (SIOP) nephroblastoma file. Histopathology. 1996;28(4):333-340. [ Links ]
54. Tomlinson GE, Breslow NE, Dome J, Guthrie KA, Norkool P, Li S, et al. Rhabdoid tumor of the kidney in the National Wilms Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23(30):7641-7645. [ Links ]
55. Haas JE, Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Hum Pathol. 1981;12(7):646-657. [ Links ]
56. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59(1):74-79. [ Links ]
57. Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB. Alterations of the hSNF5/INI1 gene in central nervous system atypical rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002;8(11):3461-3467. [ Links ]
58. Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, et al. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res. 2002;62(1):323-328. [ Links ]
59. Lee HY, Yoon CS, Sevenet N, Rajalingam V, Delattre O, Walford NQ. Rhabdoid tumor of the kidney is a component of the rhabdoid predisposition syndrome. Ped Dev Pathol. 2002;5(4):395-399. [ Links ]
60. Furchert SE, Lanvers-Kaminsky C, Juurgens H, Jung M, Loidl A, Frunwald MC. Inhibitors of histone deacetylases as potential therapeutic tools for high-risk embryonal tumors of the nervous system of childhood. Int J Cancer. 2007120(8):1787-1794. [ Links ]
61. Hoot AC, Russo P, Judkins AR, Perlman EJ, Biegel JA. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol. 2004;28(11):1485-1491. [ Links ]
62. Sigauke E, Rakheja D, Maddox DL, Hladik CL, White CL, Timmons CF, et al. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol. 2006;19(5): 717-725. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. M.Reyes Múgica
Program of Pediatric and Development Pathology
Yale University School of Medicine
430 Congress Avenue. New Haven, CT 06520-8023
Tel.: (203) 737-5097
E-mail autor: miguel.reyes@yale.edu

