










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkMedicina Intensiva
versão impressa ISSN 0210-5691
Med. Intensiva vol.32 no.4 Mai. 2008
PUESTA AL DÍA EN MEDICINA INTENSIVA: NEUROINTENSIVISMO
Estatus epiléptico
Status epilepticus
L. Corral-Ansaa, J.I. Herrero-Meseguera, M. Falip-Centellasb, M. Aiguabella-Macaub
aServicio de Medicina Intensiva.bServicio de Neurología.
Hospital Universitario de Bellvitge. L'Hospitalet de Llobregat. Barcelona. España.
Dirección para correspondencia
RESUMEN
El estatus epiléptico es una emergencia neurológica que requiere una atención inmediata. El diagnóstico y el tratamiento deben ser continuos a lo largo de los primeros minutos hasta su resolución. La causa más frecuente en pacientes epilépticos es el cambio o el incumplimiento de la medicación y en los no epilépticos son las lesiones vasculares, traumáticas, tóxicas y metabólicas. El estatus epiléptico puede ser convulsivo o no convulsivo y la monitorización electroencefalográfica continua es de gran ayuda para el diagnóstico y para valorar la respuesta al tratamiento. Las benzodiazepinas y la fenitoína o fosfenitoína son los fármacos de elección de primera y segunda línea. No existe consenso sobre los tratamientos de tercera y cuarta línea, entre los que se encuentran: fenobarbital, valproato, levetiracetam, propofol, midazolam, los barbitúricos y otros. El pronóstico dependerá de la causa, la edad, el tipo de estatus y la duración del mismo. Por ello, el tratamiento debe ser lo más precoz posible.
Palabras clave: estatus epiléptico, diagnóstico, epilepsia, tratamiento, tratamiento de emergencia.
ABSTRACT
Status epilepticus is a neurological emergency that requires prompt care. The diagnosis and treatment must be continuous from the first minutes to its resolution. The most frequent cause in epileptic patients is drug change or non-compliance and in the non-epileptic patients are cerebrovascular diseases, head trauma, drug toxicity and metabolic disturbances. Status epilepticus can be convulsive or non-convulsive and continuous electroencephalographic monitoring is useful for diagnosis and to evaluate response to treatment. Benzodiazepines and phenytoin or fosphenytoin are first-line and second-line therapy. There is no agreement on third and fourth line therapy: phenobarbital, valproate, levetiracetam, propofol, midazolam, barbiturates and others. The prognosis of status epilepticus is related to etiology, age, type and duration of the status. Thus, drug treatment for status epilepticus should be started without delay.
Key words: status epilepticus, diagnosis, epilepsy, therapeutics, emergency treatment.
Introducción
El estatus epiléptico (EE) es una emergencia neurológica que requiere una atención inmediata. La mortalidad intrínseca es del 1-7%, aunque la mortalidad global puede llegar al 20% y en casos de EE refractario hasta al 50%. Los factores que condicionan el pronóstico del EE son la edad, la duración, la etiología y la respuesta al tratamiento1. La edad y algunas causas no son modificables, pero sí que podemos actuar acortando la duración2.
El EE es una crisis epiléptica prolongada o una serie de crisis durante las cuales el paciente no recobra completamente la conciencia. Ha habido diferentes definiciones del EE en cuanto al criterio cronológico. En 1981 la Liga Internacional contra la Epilepsia3 describió el tiempo de la crisis epiléptica que llega a EE como de duración «suficiente» o «suficientemente frecuente». En 1993 la Fundación Americana de la Epilepsia4 definió el EE como crisis epiléptica que dura más de 30 minutos o dos o más crisis epilépticas subintrantes entre las cuales no existe recuperación completa de la conciencia y con una duración mayor de 30 minutos5. Una nueva definición más operativa considera como EE, en adultos o niños mayores de 5 años, cualquier actividad epiléptica de más de 5 minutos de duración, caracterizada por una crisis duradera, o dos o más crisis durante las cuales el paciente no retorna a su situación previa de conciencia5. Aunque se aceptan 30 minutos como la duración definitoria1 de un EE, consideramos que cualquier actividad epiléptica de más de 5 minutos de duración o la existencia de crisis repetidas sin recuperación del nivel de conciencia es una emergencia neurológica y debe ponerse en marcha el protocolo de tratamiento del estatus.
Se denomina EE refractario a aquél que se mantiene más de 30-60 minutos, a pesar del tratamiento adecuado con fármacos antiepilépticos de primera y segunda línea6.
La incidencia es difícil de determinar debido a las diferentes definiciones. En la población general la incidencia anual se cifra en 18-28 personas/100.000 habitantes7.
Etiología
En niños, hasta un 51% son secundarios a causas infecciosas2. En adultos, la causa más frecuente, en epilépticos previos, es la modificación del tratamiento, y en los no epilépticos, las lesiones neurológicas agudas o residuales, fundamentalmente vasculares, traumáticas, tóxicas y metabólicas (tabla 1). No se puede encontrar una causa precipitante en el 10-30% de los pacientes, sobre todo en epilépticos previos1,7.
Clínica y clasificación del estatus epiléptico
El EE se puede clasificar en estatus parcial o estatus generalizado y estos subtipos pueden dividirse a la vez en estatus no convulsivo o convulsivo; es decir, que existirían 4 tipos fundamentales de EE: parcial convulsivo, generalizado convulsivo y parcial y generalizado no convulsivo. Algunos de ellos, si no reciben tratamiento, pueden evolucionar al llamado EE sutil (que será comentado más adelante).
Estatus epiléptico parcial convulsivo
A partir de una lesión cortical focal, si las crisis son limitadas y no alteran el nivel de conciencia, se denominan crisis simples. Cursan bien con síntomas motores localizados o bien somatosensoriales, autonómicos, psíquicos o combinación de los mismos. Cuando se prolongan o repiten, dan lugar a una epilepsia parcial continua o estatus parcial simple de mejor pronóstico que otros estatus, sin precisar tratamientos muy agresivos. Sin embargo, si no se tratan y duran días o semanas, pueden provocar una necrosis cortical laminar del córtex epileptógeno.
Las crisis parciales complejas se originan en el lóbulo temporal o en estructuras límbicas, y cursan con disminución de la conciencia, mirada fija y automatismos. Suelen estar precedidas por una señal de aviso o aura (alucinaciones sensoriales, impresión de caída, disconfort abdominal, síntomas autonómicos o emocionales u otros fenómenos como el dejà vu). Una vez remitida la convulsión, el paciente puede quedar confuso unos minutos. El EE parcial complejo (EEPC) cursa con estado confusional, a veces sin otras manifestaciones salvo la actividad irritativa eléctrica frontal o temporal, que lo diferencia de otras formas generalizadas.
Estatus epiléptico generalizado convulsivo
Las crisis convulsivas generalizadas son las más frecuentes en el ámbito de Cuidados Intensivos. Se inician de forma generalizada o a partir de una crisis parcial secundariamente generalizada. En muchas ocasiones es difícil precisar si el comienzo es parcial o generalizado; sólo el electroencefalograma (EEG) puede ser diagnóstico al mostrar un foco ictal al inicio. El EE parcial convulsivo es más frecuente (7080%) y de peor respuesta terapéutica que el generalizado primario.
En su expresión típica (gran mal) cursa inicialmente con pérdida de conciencia, una fase tónica de rigidez global y una fase clónica de movimientos repetidos, sincrónicos, que van disminuyendo de frecuencia, quedando el paciente en un estado de somnolencia, confusión y amnesia de lo ocurrido (estado postictal) de pocos minutos de duración. Existen crisis generalizadas solamente con fase tónica, clónica o incluso crisis atónicas.
En el EE generalizado tonicoclónico (EEGTC), la fase tónico clónica puede durar de 30 a 60 minutos, quedando posteriormente ligeros movimientos oculares, faciales o de dedos y pasando después a una fase atónica, sin movimientos, a pesar de persistir actividad irritativa cerebral; es el llamado EE sutil, que no cursa con manifestaciones motoras pero que puede ser la fase final de un EE convulsivo.
Existen otros tipos de estatus generalizados convulsivos como el EE generalizado mioclónico (EEGM), que cursa con movimientos musculares incontrolables, rápidos, focales o generalizados, con o sin deterioro del conocimiento. A menudo se trata de parpadeo, movimientos masticatorios o sacudidas. El EEG muestra complejos punta/polipunta-onda irregulares focales, multifocales o generalizados. En adultos es secundario a encefalopatías agudas o subagudas graves (tóxico-metabólicas, hipoxia, isquemia), resistente a fármacos y de mal pronóstico. En la mayoría de los casos, excepto el de causa tóxico-metabólica, se produce en cerebros que presentan daño neuronal severo, por lo cual su tratamiento agresivo es controvertido.
Estatus epiléptico parcial o generalizado no convulsivo
Las crisis de ausencia, en las que predomina la alteración de la conciencia (pequeño mal), tienen escasas manifestaciones motoras (automatismos, parpadeo repetido, pequeñas clonias en manos, etc.). El EEG muestra puntas-onda típicas a tres hercios. El estatus de ausencias (EEGA) puede durar horas o días, pero es el único tipo que no se ha asociado con daño neuronal.
El estatus no convulsivo, oligosintomático o sutil suele aparecer al inicio de encefalopatías metabólicas graves o con lesiones estructurales subyacentes o, muy frecuentemente en Unidades de Cuidados Intensivos (UCI); suele ser consecuencia de una convulsión tónico-clónica generalizada inadvertida o parcialmente tratada. Cursa con una profunda depresión de conciencia y, a veces, sutiles manifestaciones motoras. En un 8% de pacientes con coma inexplicado se ha detectado epilepsia no convulsiva por EEG8. El diagnóstico se hace mediante el EEG, que revela descargas continuas bilaterales. Es de mal pronóstico, no sólo por la etiología, sino por el retraso en el diagnóstico.
Complicaciones cerebrales y sistémicas
En el estatus, los mecanismos compensatorios en la primera fase pueden prevenir el daño cerebral, pero a partir de los 30-60 minutos estos mecanismos fallan y se produce destrucción neuronal9. Inicialmente hay un aumento de presiones sistémicas y pulmonares y un aumento del 200-600% en el flujo cerebral. La diferencia arterio-yugular de oxígeno (DO2[a-y]) está disminuida, pero a medida que el estatus progresa, aumenta la DO2(a-y) y disminuye la presión tisular de O2. Existe un aumento de lactato cerebral (fig. 1) que se asocia a vasodilatación y aumento de la presión intracraneal. La hiperglucemia inicial mediada por catecolaminas y glucagón puede convertirse en hipoglucemia. Se produce un desacoplamiento entre la demanda y la oferta metabólica al sistema nervioso central que conduce al daño neuronal. La prolongada exposición a neurotransmisores excitatorios también contribuye a la muerte neuronal2,9. La consecuencia neuropatológica del EE es el daño neuronal selectivo, incluso irreversible, de ciertas poblaciones celulares vulnerables como las del hipocampo, la amígdala, los núcleos talámicos mediales, la corteza piriforme, las capas medias de neocórtex y las células de Purkinje del cerebelo7.
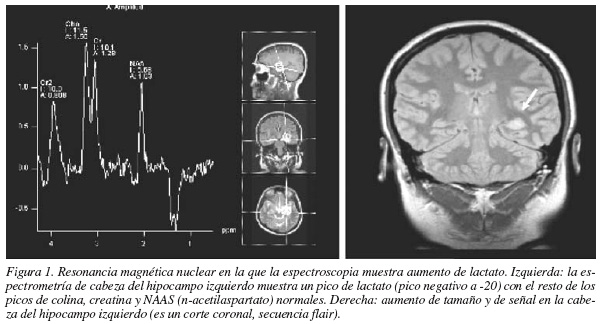
La extrema actividad muscular promueve la acidosis láctica y hay acidosis respiratoria por fracaso ventilatorio. En un 81% puede disminuir el pH por debajo de 7,3, y por debajo de 7,0 en un 35%9. La hipercaliemia, secundaria a la acidosis y al daño muscular, puede precipitar arritmias cardiacas. La hiperreactividad del sistema nervioso autónomo produce hipertermia, hipersecreción salivar y bronquial, sudoración profusa y deshidratación, que contribuye a la aparición de taquicardia, hipertensión y arritmias. También puede haber hipotensión. Puede aparecer edema pulmonar neurogénico y fracaso respiratorio, que se asocia a la hipoventilación secundaria a la hipertonía muscular y a drogas. Existe leucocitosis sistémica y discreta pleocitosis en el líquido cefalorraquídeo. La deshidratación, la rabdomiólisis y la mioglobinuria, secundaria a las contracciones musculares, pueden precipitar el fracaso renal2.
Diagnóstico
El diagnóstico del EE se basa en la presentación clínica: en adultos o niños mayores de 5 años cualquier actividad epiléptica de más de 5 minutos de duración, caracterizada por una crisis duradera, o dos o más crisis durante las cuales el paciente no retorna a su situación previa de conciencia5. Al mismo tiempo que se inician las medidas terapéuticas se deben comenzar las medidas diagnósticas para conocer la etiología7.
Ante un paciente que presenta una convulsión se debe realizar una rápida exploración clínica en busca de lesión estructural cerebral, hipertensión intracraneal, enfermedad cardiovascular como fuente embolígena, sepsis, metabolopatía u otros factores predisponentes (medicación, deprivación alcohólica o retirada de sedación). Además del análisis bioquímico se realizará un despistaje de drogas, en especial de cocaína. La tomografía axial computarizada (TAC) craneal o la resonancia magnética nuclear (RMN) nos ayudarán a localizar lesiones cerebrales, y la RMN en fase aguda es útil para la localización de la zona epileptógena (fig. 2).

La llegada de los registros electroencefalográficos digitales nos permite la monitorización continua o intermitente durante largos períodos de tiempo. Se ha detectado actividad eléctrica epiléptica subclínica hasta en un 48% y EE no convulsivo hasta en un 14% de los EE una vez que han cedido las crisis clínicas10.
Las principales indicaciones del EEG continuo son detectar crisis epilépticas no convulsivas y el EE no convulsivo, además de otras indicaciones7,11-13:
1. Es el medio de diagnóstico cuando no existe una respuesta terapéutica correcta (tipo de EE, etiología y diagnóstico diferencial).
2. Ante la recurrencia de una crisis que no se puede constatar clínicamente o en pacientes inconscientes tras el tratamiento inicial (bloqueantes neuromusculares y disociación electroclínica).
3. Para evaluar la respuesta terapéutica (desaparición de la actividad epileptiforme) y el nivel de anestesia en el EE refractario (aparición de patrón salvasupresión) (fig. 3).

Tratamiento del estatus epiléptico
Durante el período de observación inicial hay que documentar la existencia de EE y, si es posible, identificar el tipo. El EEGTC y el EEPC son emergencias que requieren diagnóstico rápido y tratamiento agresivo. El diagnóstico o el no disponer de EEG no debe retrasar el tratamiento. Cuando un tratamiento falla se debe iniciar inmediatamente el siguiente sin demora. «El tiempo es cerebro»1.
El tratamiento inicial de primer (benzodiazepinas) y segundo nivel (fenitoína) está bien protocolizado. Cuando los fármacos de primera y segunda elección fallan estamos ante un EE refractario y no existe consenso sobre los tratamientos de tercer y cuarto nivel14,15.
Medidas generales
El tratamiento del EE (fig. 4) comienza con las medidas de soporte vital, aplicables a todo paciente con deterioro del nivel de conciencia. Inmediatamente después el objetivo del tratamiento es finalizar la crisis bien con fármacos o bien identificando y tratando la causa o el factor desencadenante, corregir las complicaciones sistémicas y prevenir las recurrencias1,6,10,16,17.

1. El control de la vía aérea debe ser prioritario ycuidadoso. En la mayoría de los pacientes, a pesar de las apneas que se suceden durante las crisis, puede realizarse una adecuada ventilación y oxigenación manteniendo permeable la vía aérea, utilizando cánulas orofaríngeas o nasofaríngeas y aplicando oxigenoterapia. Se intubará cuando exista evidencia clínica o gasométrica de compromiso respiratorio.
2. Monitorizar constantes vitales y glucemia (método rápido).
3. Colocar vía venosa y realizar analítica.
4. Administrar tiamina 100 mg y glucosa al 50%50 ml (si la glucemia está baja).
5. Otros: colocar sonda nasogástrica si hay intoxicación, disminuir la hipertermia (se asocia a daño neuronal18), realizar TAC craneal, punción lumbar si se sospecha infección, dentificación y tratamiento etiológico del EE.
Tratamiento farmacológico
El tratamiento farmacológico del EE debe ser inmediato y escalonado hasta conseguir lo antes posible su resolución (tablas 2 y 3)1,6,10,14,16,17,19-22.
Primer nivel: 0-5 minutos
Están indicadas benzodiazepinas: diazepam (10-20 mg), lorazepam (2-4 mg) o clonazepam (0,5-1 mg).
Diazepam y lorazepam son de elección y de acción rápida7,14,17,23. Diazepam tarda dos minutos y lorazepam tres minutos en finalizar la crisis. La duración del efecto anticonvulsivante es mayor con lorazepam (12-24 horas) que con diazepam (15-30 minutos)11,24, aunque si se combina diazepam con fenitoína la efectividad es parecida25. Hay que repetir la dosis a los 25 minutos si la crisis no ha cedido. En España no se dispone de lorazepam endovenoso, por lo que se utiliza diazepam. Clonazepam 0,5-1 mg/min se ha utilizado eficazmente en el EE, sobre todo por ausencias o por mioclonias26-29 y podría ser una opción30.Los efectos secundarios de las benzodiazepinas son: sedación, depresión respiratoria o hipotensión. Fuera del ambiente hospitalario puede utilizarse diazepam rectal (0,5 mg/kg de preparación parenteral) o midazolam intramuscular.
Segundo nivel: 5-20 minutos
Está indicada fenitoína: 20 mg/kg a 50 mg/min. En pacientes ancianos o si hay hipotensión y/o arritmias hay que enlentecer a 25 mg/min. Tiene un efecto antiepiléptico prolongado. Se administra tras el tratamiento con una benzodiazepina o cuando ésta ha fallado. La limitación en la velocidad máxima de infusión hace que las dosis terapéuticas no puedan administrarse en menos de 20-30 minutos. Otro de sus inconvenientes es que no debe utilizarse en suero glucosado ni glucosalino porque la disminución de la concentración de sodio favorece su precipitación.
Fosfenitoína produce menos efectos secundarios durante la administración, por lo que puede administrarse en menor tiempo. En España no disponemos de fosfenitoína. Al final de este período, como opción se puede administrar una dosis adicional de fenitoína de 5-10 mg/kg. Los efectos secundarios de las hidantoínas son: hipotensión, trastornos de la conducción y repolarización cardiaca (sobre todo alargamiento del segmento QT), lesión de tejidos blandos y rara vez hipoglucemia.
Las revisiones y las guías terapéuticas basadas en la evidencia17,20,22,23 dan una recomendación grado A (extremadamente recomendable) para lorazepam, diazepam y fenitoína y una recomendación grado C (favorable pero no concluyente) para clonazepam31.
No existe consenso sobre los tratamientos de tercer y cuarto nivel17,20,22,23. Sin embargo, en la última década han aparecido varios estudios sobre eficacia y efectos adversos de valproato y muy recientemente de levetiracetam. Estos fármacos se considerarán de tercer nivel especialmente en pacientes que no sean candidatos a ingreso en la UCI o a intubación orotraqueal por su patología de base.
Valproato se administra 15-60 mg/kg en 5 minutos endovenoso, seguido de una perfusión de 1 mg/kg/h que se aumentará a 1,5 mg/kg/h en caso de haber recibido fenitoína (por inducción enzimática). La incidencia de efectos adversos sistémicos con la administración de valproato endovenoso es muy baja, aunque existe riesgo elevado de hepatotoxicidad en niños menores de dos años, en politerapia, en errores congénitos del metabolismo y en epilepsias graves acompañadas de retraso mental y de alteraciones cerebrales orgánicas. Está contraindicado en las alteraciones de la coagulación, hepatopatía y pancreatopatía severas32. Un estudio comparativo reciente ha mostrado una eficacia superior de valproato respecto a fenitoína en EE convulsivos33. Valproato es de primera elección en EEGM o EEGA.
Levetiracetam es un nuevo fármaco antiepiléptico comercializado en España desde 2001 y recientemente disponible por vía endovenosa. Derivado de pirrolidona, tiene un mecanismo de acción diferente al resto de fármacos antiepilépticos (no afecta a los receptores del glutamato ni del ácido gamma aminobutírico y se une a la proteína SVA2 de las vesículas sinápticas), con buena biodisponibilidad. Se utiliza como monoterapia en crisis parciales y como terapia concomitante en otros tipos de epilepsia. No puede administrarse en la misma vía que fenitoína porque precipita, pero sí puede administrarse con valproato o benzodiazepinas. Existen casos de EE refractarios tratados con levetiracetam 1.000 mg en 15 minutos endovenoso con buen resultado34.
Tercer nivel: 30 minutos
Si persisten convulsiones será necesario la intubación, la ventilación mecánica y la monitorización hemodinámica.
Fenobarbital, a pesar de su eficacia, cada vez se utiliza menos como antiepiléptico en los países desarrollados debido a sus efectos adversos y neurotóxicos (alteraciones del comportamiento, cognitivos y depresión)35. En el EE se ha utilizado a dosis de 20 mg/kg a ritmo de 100 mg/minuto, recomendación grado C (favorable pero no concluyente)2,7,9,11,12,14,17,20,21,23. En caso de administrarlo, hay que mantener fenitoína y fenobarbital durante la fase aguda. Los efectos secundarios son: sedación, depresión respiratoria, hipotensión, neurotoxicidad o rash. Una opción al final de este periodo es una dosis adicional de fenobarbital de 5-10 mg/kg.
Cuarto nivel: 60-65 minutos
Se indica anestesia general con barbitúricos de acción rápida, propofol o midazolam. Las dosis requeridas para el control del EE pueden producir efectos secundarios hemodinámicos y sistémicos graves. Se deben de conocer y utilizar en UCI. Se requiere monitorización continua/frecuente de EEG y si se produce hipotensión administrar volumen y utilizar vasopresores adecuadamente. El objetivo con estos fármacos tiene como finalidad llegar a la salva-su-presión en el EEG, ya que consigue mejor el cese del estatus y evita la reaparición de las crisis36.
1. Tiopental sódico: 3 mg/kg en 3-5 minutos se-guido de 2 mg/kg/h. Si recurren las convulsiones administrar 50-100 mg en bolus y aumentar la velocidad en 0,5-1 mg/kg/h hasta 5 mg/kg/h.
2. Pentobarbital: 5-20 mg/kg a 0,2-0,4 mg/kg/minseguido de 2,5 mg/kg/h.
3. Propofol: existe controversia sobre su posibleacción proconvulsivante, pero se ha mostrado eficaz en el tratamiento del EE refractario. Inicialmente 1-2 mg/kg en 5 minutos, seguido de una perfusión entre 2-10 mg/kg/h.
4. Midazolam: 0,2 mg/kg en bolus, seguido de 0,1-2,0 mg/kg/h. Se ha utilizado para el EE refractario de forma eficaz37.
Los resultados en el EE refractario con midazolam, propofol o barbitúricos son similares y lo que más influye en la evolución es la causa del EE19,38. La preferencia de utilización de los fármacos de tercer o cuarto nivel varía en los diferentes esquemas publicados1,7,9,17,19,20,21, aunque en la práctica14 se sigue el orden explicado.
Minuto 80
En este momento las posibilidades de recuperación satisfactorias son muy reducidas. Se deben reevaluar los pasos anteriores: errores en el diagnóstico, en la administración de fármacos, en la identificación y la corrección de causas o complicaciones. Se debe considerar la asociación de otros fármacos, aunque su utilidad sea incierta, como: carbamacepina, topiramato, clormetiazol, lidocaína, paraldehído, primidona, etosuximida, agentes inhalantes (isofluorano, óxido nitroso y halotano), ketamina y otros (cloracepato, esteroides, etomidato, gabapentina, lamotrigina, piridoxina, magnesio y tiagabina)1,7,21. Los relajantes musculares sólo deben utilizarse como último recurso en convulsiones intratables cuando exista acidosis, apnea o contracciones musculares con amenaza para la vida39.
También se han utilizado ocasionalmente tratamientos no farmacológicos como el electroshock40 y en casos de EE parciales motores que hayan producido una epilepsia parcial continua, tratamiento quirúrgico con resecciones transpiales múltiples41.
Duración del tratamiento
El objetivo del tratamiento debe ser la finalización de la crisis epiléptica clínica o electroencefalográfica. La mayoría de las veces se disminuye el tratamiento entre 24 y 48 horas tras conseguir el control del estatus15,17. La retirada de todos los fármacos anestésicos debe siempre realizarse de forma gradual (20% cada 6 horas ) y cuando se tengan niveles terapéuticos en sangre de los fármacos de segunda línea (fenitoína o valproato o ambos). Se acepta conseguir la salva-su-presión, pero existe controversia sobre si es mejor conseguir el cese de las crisis eléctricas o si hay que alcanzar la salva-supresión, o llegar a aplanamiento del EEG global35. La sedación se retira a las 24 horas del cese de las crisis y si aparecen convulsiones se vuelve a administrar 24 horas más1.
Pronóstico
La mortalidad en el EE está alrededor del 20% (oscila entre el 7 y el 50%)2,7,9,11,19. La mortalidad en el EE refractario es del 43% a los 30 días y en edades avanzadas puede ser del 76%16. El pronóstico depende sobre todo de la causa, además de la presentación clínica, la edad y la duración del EE1,7,16.
Las causas agudas, tóxico-metabólicas, infecciosas, neoplásicas, hipóxicas y de accidentes cerebrovasculares tienen peor pronóstico que los epilépticos previos en los que hay cambio de medicación o en enólicos7,9. El EEGA tiene buen pronóstico, el EEPC es de mal pronóstico y el EEGM posthipoxia tiene muy mal pronóstico7,11. La duración del EE influye en la evolución: a partir de los 30 minutos la mortalidad es mucho mayor9. El EE también puede producir secuelas neuropsicológicas, cognitivas y de memoria9.
Declaración de conflicto de intereses
Los autores han declarado no tener ningún conflicto de intereses.
Bibliografía
1. Chen JWY, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol. 2006;5:246-56. [ Links ]
2. Bassin S, Smith TL, Bleck TP. Clinical review: Status epilepticus. Crit Care. 2002:6:137-42. [ Links ]
3. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electrographic classification of epileptic seizures. Epilepsia. 1981;22:489-501. [ Links ]
4. Dodson WE, DeLorenzo RJ, Pedley TA. For the epilepsy foundation of America's working group on status epilepticus. Treatment of convulsive status epilepticus. JAMA. 1993;270:854-9. [ Links ]
5. Lowenstein DH, Bleck T, Mcdonald RL. It's time to revise the definition of status epilepticus. Epilepsia. 1999;40:120-2. [ Links ]
6. Claassen J, Hirsch LJ, Emerson RG. Continuous EEG monitoring and midazolam infusion for refractory nonconvulsive status epilepticus. Neurology. 2001;57:1036-42. [ Links ]
7. Tejeiro J, Gómez-Sereno B. Status epilepticus. Rev Neurol. 2003:36:661-79. [ Links ]
8. Towne AR, Waterhouse EJ, Boggs JG. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology. 2000;54:340-5. [ Links ]
9. Chapman MG, Smith M, Hirsch NP. Status epilepticus. Anaesthesia. 2001;56:648-59. [ Links ]
10. DeLorenzo RJ, Waterhouse EJ, Towne AR, Boggs JG, Ko D, DeLorenzo GA, et al. Persitent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia. 1998;39:833-40. [ Links ]
11. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338:970-6. [ Links ]
12. Sirven J, Waterhouse E. Management of status epilepticus. Am Fam Physician. 2003;68:469-76. [ Links ]
13. Hirsch LJ. Continuous EEG Monitoring in the Intensive Care Unit: an overview. J Clin Neurophysiol. 2004;21:332-40. [ Links ]
14. Claassen J, Hirsch LJ, Mayer SA. Treatment of status epilepticus: a survey of neurologists. J Neurol Sci. 2003;211:37-41. [ Links ]
15. Holtkamp M, Masuhr F, Harms L, Einhäulpl KM, Meierkord H, Buchheim K. The management of refractory generalised convulsive and complex partial status epilepticus in three European countries: a survey among epileptologists and critical care neurologists. J Neurol Neurosurg Psychiatry. 2003;74:1095-9. [ Links ]
16. Marik PE, Varon J. The management of status epilepticus. Chest. 2004;126:582-91. [ Links ]
17. Meierkord H, Boon P, Engelsen B, Göcke K, Shorvon S, Tinuper P, et al. EFNS guideline on the management of status epilepticus. Eur J Neurol. 2006;13:445-50. [ Links ]
18. Simon RP. Physiologic consequences of status epilepticus. Epilepsia. 1985;26 Suppl 1:S58-66. [ Links ]
19. Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol or midazolam: a systematic review. Epilepsia. 2002;43:146-53. [ Links ]
20. Serrano-Castro PJ, Casado-Chocán JL, Mercadé-Cerdà JM, Altuzarra-Corral A, Rufo-Campos M, Moreno-Alegre V, et al. Guía terapéutica en epilepsia de la Sociedad Andaluza de Epilepsia 2005: III. Tratamiento antiepiléptico en situaciones especiales. Rev Neurol. 2005;40:683-95. [ Links ]
21. Lowenstein DH. The management of refractory status epilepticus: an update. Epilepsia. 2006;47 Suppl 1:35-40. [ Links ]
22. Prasad K, Al-Roomi K, Krishnan PR, Sequeira R. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;19:CD003723. [ Links ]
23. Walker M. Status epilepticus: an evidence based guide. BMJ. 2005;331:673-7. [ Links ]
24. Aldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, et al. A comparison of lorazepam, diazepam and placebo for the treatment of out-of-hospital status epilepticus. N Eng J Med. 2001;345:631-7. [ Links ]
25. Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Eng J Med. 1998;339: 792-8. [ Links ]
26. Sorel L, Mechler L, Harmant J. Comparative trial of intravenous lorazepam and clonazepam in status epilepticus. Clin Ther. 1981;4:326-36. [ Links ]
27. Singh AN, Le Morvan T. Treatment of status epilepticus with intravenous clonazepam. Prog Neuropsychopharmacol Biol Psychiatry. 1982;6:539-42. [ Links ]
28. Rossetti AO, Reichhart MD, Schaller MD, Despland PA, Bogousslavsky J. Propofol treatment of refractory status epilepticus: a study of 31 episodes. Epilepsia. 2004;45:757-63. [ Links ]
29. Baykan B, Gokyigit A, Gurses C, Eraksoy M. Recurrent absence status epilepticus: clinical and EEG characteristics. Seizure. 2002;11:310-9. [ Links ]
30. Rey E, Treluyer JM, Pons G. Pharmacokinetic optimization of benzodiazepine therapy for acute seizures. Focus on delivery routes. Clin Pharmacokinet. 1999;36:409-24. [ Links ]
31. Brainin M, Barnes M, Baron JC, Gilhus Ne, Hughes R, Selmaj K, et al; Guideline Standards Subcommittee of the EFNS Scientific Committee. Guidance for the preparation of neurological management guidelines by EFNS scientific task forces – revised recomendations 2004. Eur J Neurol. 2004;11:577-581. [ Links ]
32. Adín J, Arteaga R, Herranz JL, Armijo JA. Utilización del valproato por vía intravenosa. Rev Neurol. 1999;29:744-53. [ Links ]
33. Misra UK, Patel R, Kalita J. Sodium valproate vs phenytoin in status epilepticus. A pilot study. Neurology. 2006;67:340-2. [ Links ]
34. Schulze-Bonhage A, Hefft S, Oehl B. Termination of complex partial status epilepticus by intravenous levetiracetam- a case report. J Neurol Neurosurg Psychiatry. 2007. [ Links ]
35. Kwan P, Brodie MJ. Phenobarbital for the treatment of epilepsy in the 21st century: a critical review. Epilepsia. 2004;45: 1141-9. [ Links ]
36. Krishnamurthy KB, Drislane FW. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia. 1999;40:759-62. [ Links ]
37. Ulvi H, Yoldas T, Múngen B, Yigiter R. Continuous infusion of midazolam in the treatment of refractory generalized convulsive status epilepticus. Neurol Sci. 2002;23:177-82. [ Links ]
38. Rossetti AO, Logroscino G, Bromfield E. Refractory status epilepticus. Arch Neurol. 2005;62:1698-702. [ Links ]
39. Nirmel KN, Kofke WA, Ropper AH. Estado de mal epiléptico. En: Kofke WA, Levy JH, editores. Procedimientos de cuidados intensivos postoperatorios del Massachusetts General Hospital. 1.ª edición. Barcelona: Salvat Editores; 1990. p. 431-43. [ Links ]
40. Cline JS, Roos K. Treatment of status epilepticus with electroconvulsive therapy. J ECT. 2007;23:30-2. [ Links ]
41. Molyneux PD, Barker RA, Thom M, Paesschen WV, Harkness WF, Duncan JS. Successful treatment of intractable epilepsia partialis continua with multiple subpial transections. J Neurol Neurosurg Psychiatry. 1998;65:137-8. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dra. L. Corral Ansa.
Servicio de Medicina Intensiva.
Hospital Universitario de Bellvitge.
Feixa Llarga, s/n.
08907 L'Hospitalet de Llobregat. Barcelona. España.
Correo electrónico: 30130lca@comb.es
Manuscrito aceptado el 25-I-2008.

