










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Intensiva
versión impresa ISSN 0210-5691
Med. Intensiva vol.33 no.3 abr. 2009
PUESTA AL DÍA: NEUROINTENSIVISMO
Patología neuromuscular en cuidados intensivos
Neuromuscular abnormalities in critical illness
R. Amaya Villar, J. Garnacho-Montero y M.D. Rincón Ferrari
Servicio de Cuidados Críticos y Urgencias. Unidad de Cuidados Intensivos. Hospitales Universitarios Virgen del Rocío. Sevilla. España.
Dirección para correspondencia
RESUMEN
La patología neuromuscular en los pacientes críticos ha comenzado a ser objeto de un importante número de estudios en los últimos años, si bien aún quedan muchas lagunas en el conocimiento de su etiología, patogenia, tratamiento y pronóstico. Dentro de esta patología debemos distinguir dos grandes grupos. En el primero, la debilidad muscular aparece antes del ingreso en UCI y es posible identificar una causa conocida. El síndrome de Guillain-Barré y la miastenia grave son las dos entidades que con mayor frecuencia requieren ser atendidas en nuestras unidades. En el segundo grupo, la debilidad muscular se adquiere en la UCI, en pacientes sin enfermedad neuromuscular previa, y es secundaria a la gravedad de la enfermedad que originó su ingreso en esta unidad y/o al tratamiento empleado. La polineuropatía del paciente crítico (PPC) es, de todas ellas, la entidad más precisamente definida y de la que conocemos mejor sus características clínicas, diagnóstico y pronóstico; no obstante, aún quedan muchas sombras en cuanto a su etiopatogenia. Las alteraciones de la placa neuromuscular y sobre todo la miopatía, que frecuentemente coexiste con la PPC, son las otras complicaciones del sistema nervioso periférico que se desarrollan en pacientes críticos. Los avances en el conocimiento de estas afecciones podrían tener un importante impacto, sobre todo para el desarrollo de intervenciones terapéuticas y preventivas efectivas que mejoren el pronóstico de estos pacientes.
Palabras clave: Debilidad muscular. Paciente crítico. Etiopatogenia. Tratamiento. Pronóstico.
ABSTRACT
The spectrum of neuromuscular disease encountered in today's intensive care units (ICU) has evolved over the last few decades. However, in spite of many studies on neuromuscular disorders complicating critical illness as well as its epidemiology, etiology, treatment and prognosis, several key areas remain unclear. Two main groups are found among these neuromuscular abnormalities. The first group includes primary neuromuscular disorders present on admission to the ICU in which a possible etiology can be identified. Guillain-Barré syndrome and myasthenia gravis are two of the most common diseases admitted to ours units. In the second group, weakness is acquired in the ICU in the absence of preexisting neuromuscular disease. It is believed to reflect illnesses or treatments occurring in the ICU. Critical illness polyneuropathy (CIP) is the most clearly defined neuromuscular complication in this group. However, although we have better knowledge of its clinical, diagnosis, and prognosis features, its pathophysiological substrate has not been fully elucidated. Neuromuscular junction defects and specially myopathies, that frequently coexist with CIP, are the others main causes of acquired weakness in critically ill patients. Advances in understanding of these neuromuscular disorders could have an important impact in terms of developing effective preventive and therapeutic interventions that could help to improve the poor prognosis of these patients.
Key words: Muscle weakness. Critical illness. Pathophysiology. Treatment. Prognosis.
Síndrome de Guillain-Barré (SGB)
El SGB es una polirradiculoneuropatía aguda inflamatoria ascendente que generalmente tiene una remisión espontánea. Sin embargo, a veces puede causar insuficiencia respiratoria, lo cual obliga al ingreso en la unidad de cuidados intensivos (UCI)1.
Incidencia y etiopatogenia
Se ha descrito una incidencia de 1-4 casos/100.000 hab/año, y es más frecuente en varones, con una relación 2:1.
La etiología es desconocida, aunque se acepta la intervención de un mecanismo inmunitario no bien entendido. Según esta respuesta inmunitaria afecte a la mielina o al axón de forma primaria, se pueden diferenciar cuatro subtipos, dos con alteración primaria de la mielina: la polineuropatía desmielinizante inflamatoria aguda (la forma más frecuente en Europa y Norteamérica [95%], y a la que nos referiremos en esta revisión) y el síndrome de Miller-Fisher; y dos con afección primaria del axón: neuropatía axónica motriz sensitiva aguda y la neuropatía axónica motriz aguda2.
El papel de la inmunidad celular no está aún bien determinado. Además, por otro lado, se ha descrito la existencia de anticuerpos contra los nervios periféricos y depósito de inmunoglobulinas en la mielina de los nervios periféricos3.
No se conocen los antígenos que ponen en marcha esta respuesta, aunque se postula que sean infecciosos, dado que dos tercios de los pacientes tienen un cuadro catarral o diarreico en las 4 semanas previas4. Se han postulado diversos agentes: virus como citomegalovirus, virus de Epstein-Barr, varicela-zoster o VIH; bacterias como Campylobacter jejuni (el antecedente más frecuente) o Mycoplasma, aunque en otras ocasiones no se identifica el microorganismo; también vacunas como la de la rabia o la polio oral. En otros casos existen antecedentes no infecciosos como linfoma o lupus eritematoso sistémico3.
Manifestaciones clínicas
Los pacientes suelen aquejar un cuadro inespecífico respiratorio o de diarreas antes de los síntomas neurológicos, los cuáles comienzan con dolor y parestesias distales seguidas de debilidad en miembros inferiores que dificulta la deambulación. El déficit motor asciende, con arreflexia y de forma generalmente simétrica, afectando a extremidades superiores, músculos faciales y de orofaringe, lo cual ocurre en 2-4 semanas. Puede haber afección de los pares craneales, y el nervio facial es el más frecuentemente involucrado. También puede haber afección bulbar y de los nervios motooculares. Existen casos de evolución rápida en los que se instaura parálisis generalizada en tan sólo unos días5.
En un 25% de los casos se afectan los músculos respiratorios, lo que hace necesaria la ventilación mecánica (VM)3,6. Un 8-16% de los pacientes presentan recaídas tras la mejoría inicial.
Es frecuente la disfunción del sistema nervioso autónomo7. La forma de presentación es muy variada (tabla 1). También se ha asociado a hiponatremia por síndrome de secreción inadecuada de vasopresina (ADH)8.

Diagnóstico
El diagnóstico se basa en los hallazgos clínicos (comentados previamente), el análisis del líquido cefalorraquídeo (LCR) y los hallazgos neurofisiológicos.
Los cambios característicos del LCR aparecen tras la primera semana y consisten en elevación de las proteínas totales sin incremento de la celularidad, lo que también se conoce como disociación albuminocitológica3.Raramente, el LCR puede permanecer normal o mostrar pleocitosis ligera (50 linfocitos/μl).
El estudio neurofisiológico (ENF) es la mejor prueba confirmatoria y permite diferenciar las variantes axonales y desmielinizantes9. Requiere encontrar al menos dos nervios individuales explorados con datos concordantes con una polineuropatía desmielinizante: reducción de la velocidad de conducción nerviosa, latencias distales aumentadas y bloqueo de conducción motriz parcial. No obstante, en un 5% de los casos el patrón es de una degeneración axonal3. El mejor momento para la realización del ENF aún no ha sido descrito, pero se recomienda realizarlo tan pronto como sea posible tras el inicio de la clínica, y repetirlo 1-2 semanas después si el estudio inicial no fue diagnóstico o no permitió una adecuada clasificación neurofisiológica. La electromiografía puede ayudar midiendo la pérdida axonal y dar una aproximación del pronóstico3.
Una vez realizado el diagnóstico de neuropatía periférica aguda, el SGB es la etiología más frecuente, aunque no la única. El diagnóstico diferencial se debe realizar con los trastornos neuromusculares que los pacientes críticos pudieran tener antes de su ingreso en UCI (tabla 2).

En el SGB la histología revela inflamación de los nervios periféricos tanto sensitivos como motores. Las lesiones son multifocales y se aprecia infiltración de linfocitos y posteriormente de macrófagos con pérdida de la vaina de mielina. El daño axonal se supone es secundario en el 95% de los casos. En ocasiones se aprecia degeneración de las motoneuronas del asta anterior y de las columnas dorsales secundarias a las lesiones de los nervios periféricos.
Tratamiento
Es fundamental llevar a cabo un manejo multidisciplinario para la prevención y el tratamiento de las complicaciones potencialmente fatales de esta enfermedad.
La insuficiencia respiratoria es la causa que casi de forma constante justifica el ingreso en la UCI y hace preciso generalmente usar la VM6. Las formas graves de la enfermedad, es decir, aquellas con rápida progresión, parálisis bulbar y disfunción autonómica, deben ser estrechamente monitorizadas por el posible desarrollo de arritmias cardíacas, que pueden incluso llevar al fallecimiento del paciente6. El tratamiento de la disautonomía es sintomático y a veces requiere el empleo de hipotensores y sedación ante las crisis hipertensivas o, por contra, el empleo cuidadoso de aminas para tratar la hipotensión. En ocasiones, episodios de bradicardia severa y bloqueos de conducción que no revierten con atropina hacen preciso colocar un marcapasos transitorio7,9.
En los últimos años se han producido importantes avances en la inmunoterapia en el SGB, a pesar de lo cual persisten diferentes aspectos sin dilucidar. La plasmaféresis (PF) se considera el tratamiento de elección, ya que se ha demostrado que reduce el tiempo de conexión a VM y de recuperación motriz general10-12. Se piensa que el efecto beneficioso de la PF en esta entidad está condicionado por la eliminación de anticuerpos específicos contra la mielina junto con otras proteínas solubles del suero. El número de sesiones no está claramente definido, pero se recomienda cinco realizadas en días alternos12. Tras la mejoría inicial, se produce recaída en un 10-20% de los pacientes. En estos casos se puede recurrir a sesiones adicionales de PF o al tratamiento con inmunoglobulinas intravenosas (IGIV)13.
El empleo de IGIV a altas dosis (0,4 g/kg/día) durante 5 días consecutivos se ha comunicado tan efectivo como la PF en cuanto a la mejoría de los síntomas14. El mecanismo de acción de la IGIV probablemente sea multifactorial, con bloqueo de los receptores e interferencia en la activación del complemento y la regulación de las células T. Hughes et al15 no observaron diferencias en la duración de la VM, muerte o discapacidad residual entre los dos tratamientos (PF o IGIV). La combinación de PF e IGIV no ha demostrado ventajas adicionales16.
El uso de corticoides ha sido controvertido, pero en la actualidad no se recomienda ya que ningún estudio ha sido capaz de probar que influya en el curso de la enfermedad17.
Recientemente se ha postulado como opción terapéutica la filtración de LCR (licuoféresis) para eliminar factores humorales potencialmente desmielinizantes del LCR, lo que acortaría el curso de la enfermedad. Los resultados son prometedores, aunque todavía son necesarios estudios clínicos amplios, controlados y aleatorizados para obtener conclusiones definitivas18 (tabla 3).
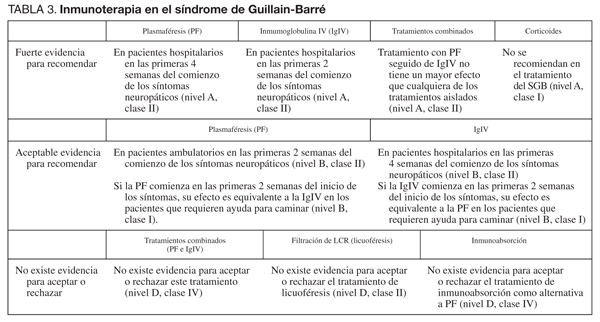
El tratamiento analgésico, el soporte psicoemocional y la fisioterapia respiratoria son imprescindibles en estos pacientes6. Un programa de rehabilitación multidisciplinario es tan importante como la inmunoterapia, ya que un alto porcentaje de estos pacientes van a presentar fatiga persistente, probablemente secundaria a la pérdida de axones, y pueden experimentar una notable mejoría con un determinado programa de ejercicios3,6.
Pronóstico
En general, más de dos tercios de los pacientes presentan una recuperación completa en semanas o meses. La mortalidad oscila entre un 4 y un 15% de los pacientes, y un 4% de los casos precisan soporte ventilatorio de por vida.
La edad, el encamamiento o la necesidad de VM al inicio de la enfermedad, la ausencia de respuesta motriz y la afección axonal inicial observada en el ENF se han identificado como factores de mal pronóstico19,20. Visser et al20 demostraron que en los pacientes con episodios diarreicos previos o con infección por C. jejuni se desarrolla una enfermedad más severa y una recuperación más lenta.
Por otro lado, se ha observado, incluso en pacientes con buena recuperación, la existencia de debilidad residual y pérdida de las unidades motrices en el ENF, que podrían explicar la fatiga persistente en estos pacientes21.
Son necesarios más estudios para identificar mejores regímenes terapéuticos y/o nuevas estrategias terapéuticas, especialmente en este subgrupo de mayor severidad, para disminuir la elevada morbimortalidad asociada aún a esta enfermedad20.
Miastenia grave
Es una enfermedad que afecta a la unión neuromuscular y que se caracteriza por presentar debilidad y fatigabilidad de los músculos esqueléticos.
Incidencia y etiopatogenia
Se ha descrito una incidencia alrededor de 3 casos/10.000 personas. Es más común en mujeres jóvenes y varones de edad avanzada, aunque puede presentarse a cualquier edad.
La miastenia grave (MG) es una enfermedad auto-inmunitaria adquirida en la que se producen anticuerpos contra los receptores nicotínicos de acetilcolina (ACRA) de la membrana postsináptica en la unión neuromuscular. Ello trae como consecuencia una reducción en el número de receptores, lo que hace imposible mantener la contracción muscular22. Se han descrito tres mecanismos mediante los cuales los ACRA reducen el número de receptores: bloqueo del receptor, destrucción del receptor vía activación del complemento y aceleración de la endocitosis del receptor. Se desconoce el agente que pone en marcha esta agresión, pero existe suficiente evidencia de que el timo desempeña un papel importante en la patogenia de esta enfermedad. El timo contiene células semejantes a los miocitos con receptores de acetilcolina en su superficie. Estas células, vulnerables al daño inmunitario, liberarían la proteína sensibilizante de estímulo antigénico. Los linfocitos del timo (linfocitos T) estimularían a los linfocitos B, los cuales, a su vez, producirían los ACRA. Se ha descrito en el 75% de los casos una hiperplasia del timo, correspondiendo en un 10% a un tumor tímico23.
Por otro lado, se sabe que la actividad repetida acaba disminuyendo la cantidad de acetilcolina liberada (agotamiento presináptico). Además, la activación de las fibras musculares es cada vez menor por un impulso sucesivo (fatiga miasténica). Estos mecanismos explicarían el aumento de la fatiga tras el ejercicio y la estimulación decreciente en el electromiograma.
Manifestaciones clínicas
La debilidad y la fatiga musculares son los síntomas cardinales. Los músculos más frecuentemente afectados son los extraoculares, manifestándose por diplopía y ptosis palpebral, los de la lengua y deglución, así como los proximales de las extremidades. Esta predilección por ciertos grupos musculares se ha atribuido a varios factores, uno de los cuales es la diferencia de temperatura entre ellos.
La clínica puede variar desde formas leves, con afección exclusivamente ocular, a las formas más graves con debilidad generalizada e insuficiencia respiratoria.
La insuficiencia respiratoria es la principal causa de ingreso en UCI, y requiere generalmente la conexión al respirador. En ocasiones, el inicio de los síntomas coincide con una enfermedad sistémica (especialmente hipotiroidismo o hipertiroidismo), con infecciones intercurrentes, fiebre, agotamiento físico o emocional y embarazo o puerperio. La debilidad muscular mejora con el frío. Por el contrario, ciertos medicamentos como los antibióticos aminoglucósidos, tetraciclinas, antiarrítmicos, bloqueadores beta, fenotiacidas y magnesio empeoran la función muscular en estos pacientes24.
Un 15-20% de los pacientes sufren una crisis miasténica, precipitada habitualmente por una infección respiratoria interrecurrente (bacteriana o viral) y caracterizada por afección respiratoria que requiere VM25. Por otro lado, se debe excluir la posibilidad de que la crisis sea causada por un tratamiento excesivo de la medicación anticolinérgica (crisis colinérgica). En estos casos, se aprecian efectos colinérgicos (miosis, bradicardia, salivación, vómitos y diarrea, entre otros) y al realizar el test del Tensilón® no se observa mejoría e incluso aumenta la debilidad.
Diagnóstico
La mayoría de los pacientes que ingresan en UCI ya tienen previamente establecido el diagnóstico. En caso contrario, se trata de una insuficiencia respiratoria con afección de pares craneales, por lo que debemos establecer el diagnóstico diferencial con el botulismo, en el que habrá antecedentes epidemiológicos y de cuadro gastrointestinal, polimiositis, esclerosis lateral amioatrófica y síndrome miasteniforme de Eaton-Lambert.
Para el diagnóstico se emplea el test del Tensilón®, que consiste en la administración de 10 mg de edrofonio (Tensilón®) intravenoso en 30 s, con lo cual mejorará ostensiblemente la fuerza muscular. Se debe disponer de una jeringa con atropina para controlar los síntomas gastrointestinales o, en casos raros, de bradicardia e hipotensión. Esta prueba es de gran utilidad en pacientes con ptosis o debilidad de los músculos extraoculares y tiene una sensibilidad del 80-95% en pacientes con MG ocular (MGO)24. Se han descrito falsos positivos en pacientes con enfermedad de neurona motriz, síndrome de Guillain-Barré, síndrome miasténico, tumores de la hipófisis y neuropatías oculares diabéticas.
En la MG los estudios de conducción nerviosa son normales, pero en el electromiograma (EMG) se aprecia una reducción de la amplitud del potencial de acción de la unidad motriz, con un decremento a la estimulación repetitiva. Por el contrario, el síndrome miasteniforme de Eaton-Lambert o trastornos metabólicos tales como la hipermagnesemia o la hipocalcemia muestran una facilitación tras la estimulación repetitiva. Esta prueba no es específica de la MG, ya que puede ser positiva en otras enfermedades neuromusculares. El electromiograma de fibra aislada (EMGFA) es la prueba más sensible para estudiar la transmisión neuromuscular. Aproximadamente un 90% de los pacientes con MG leve y un 60% con MGO presentan un EMGFA anormal, por lo que podría considerarse una herramienta útil para el diagnóstico precoz en estos pacientes24.
La presencia de ACRA es muy específico de la MG y se encuentran en un 75% de los casos. Es prácticamente constante en los pacientes con timoma y no se suele detectar en los casos limitados a la musculatura ocular. Aunque los falsos positivos son raros, se han observado en pacientes con lupus eritematoso y con enfermedades hepáticas autoinmunitarias24.
Se recomienda la realización de una tomografía computarizada o una resonancia magnética de tórax en todo paciente diagnosticado de MG para el estudio del timo.
Tratamiento
El tratamiento de la MG ha avanzado en los últimos años, especialmente debido a la disponibilidad de nuevos fármacos; sin embargo, todavía se discute cuál es el tratamiento más eficaz en estos pacientes.
Los fármacos anticolinesterásicos constituyen la base fundamental en el manejo de esta entidad. Sin embargo, son útiles para el tratamiento sintomático, pero no influyen en la patogenia de la enfermedad. El bromuro de piridostigmina (Mestinón®) es el agente oral más utilizado. No existe una pauta establecida de tratamiento porque las respuestas varían entre pacientes y en un mismo enfermo. Se administra cada 4-6 h y la dosis diaria oscila entre 180 y 540 mg. La sobredosis de anticolinesterásicos puede producir un aumento de la debilidad y otros efectos secundarios muscarínicos como diarrea, espasmos abdominales, sialorrea (hipersalivación) o náuseas. Para evitar estos síntomas digestivos, son útiles la atropina-difenoxilato y la loperamida.
Los corticoides producen mejoría en el 80% de los casos. Se recomienda metilprednisolona 80-240 mg/ día por vía intravenosa o prednisona oral 60-180 mg/ día. Es importante un control minucioso del paciente, ya que puede producirse una exacerbación de la debilidad muscular 1-2 semanas tras comenzar la administración de prednisona. Esta recaída, en la mayoría de los casos, se controla con Mestinón® o PF, aunque excepcionalmente puede llevar a insuficiencia respiratoria y necesidad de VM. Algunos pacientes requieren dosis bajas de forma crónica para evitar recaídas26.
La PF consigue disminuir por un corto plazo la concentración de anticuerpos anticolinesterásicos, con mejoría clínica en muchos pacientes. Se recomiendan cinco sesiones en días alternos. Actualmente, las indicaciones aceptadas son como medida temporal en pacientes graves, previa a la cirugía (p. ej., para la timectomía) y en la crisis miasténica27.
Las indicaciones de la IGIV son las mismas que las de la PF, con la ventaja de que no requiere un equipo especial ni una vía venosa de gran calibre. Se desconoce su mecanismo de acción, pero no se ha observado una disminución de la concentración de anticuerpos anticolinesterásicos. La dosis administrada es de 0,4 g/kg/día durante 5 días consecutivos y los beneficios son transitorios. Actualmente se reserva para casos que no responden a PF o cuando no sea técnicamente posible su realización28.
Finalmente, el uso de fármacos inmunosupresores a largo plazo parece ser eficaz, aunque hay datos contradictorios en los pocos estudios llevados a cabo en poblaciones heterogéneas y con un número escaso de pacientes. Recientemente, Hart et al29 describieron una mejoría significativa en la MG generalizada con ciclosporina como monoterapia y/o corticoides, o con ciclofosfamida más corticoides. Por el contrario, no se observó ningún beneficio en el tratamiento con azatioprina (en monoterapia o con esteroides), micofelonato mofetilo (en monoterapia o con esteroides o ciclosporina) o tacrolimus (con corticoides o PF). Ante estos hallazgos, son necesarios futuros estudios en un intento de aclarar el tratamiento inmunosupresor más adecuado en estos pacientes.
La timectomía está indicada en todos los pacientes con miastenia generalizada entre la pubertad y los 55 años como mínimo, y aún no hay consenso en otros grupos de edad o en pacientes con debilidad limitada a la musculatura ocular. En general, un 85% de los pacientes mejoran con la timectomía; un 35% entra en remisión, sin necesitar tratamiento farmacológico, y un 50% reduce los requerimientos de medicación30.
El tratamiento de la crisis miasténica debe realizarse en una UCI, por un equipo con experiencia en el tratamiento de este tipo de crisis, de la insuficiencia respiratoria y de las complicaciones infecciosas y la terapia hidroelectrolítica25.
Pronóstico
A pesar de los avances terapéuticos y del manejo cada vez más especializado, presenta una mortalidad de un 3-8%25.
No hay cura, pero es posible una remisión a largo plazo. En la actualidad, casi todos los pacientes pueden reanudar una vida normal con un tratamiento adecuado. La calidad de vida de estos pacientes dependerá tanto de la severidad de la enfermedad como de los efectos secundarios del tratamiento empleado, sobre todo de la terapia inmunosupresora administrada a largo plazo24.
Polineuropatía del paciente crítico
La polineuropatía del paciente crítico (PPC) es una degeneración axonal primaria de fibras motrices y sensitivas que se acompaña de degeneración de las fibras musculares como resultado de la denervación aguda que sufren dichas fibras del músculo estriado31. Ocurre en pacientes críticos, especialmente los que contraen síndrome de respuesta inflamatoria sistémica (SRIS) y sepsis grave con síndrome de disfunción multiorgánica (SDMO). Generalmente, la PPC coexiste con la miopatía del paciente crítico, por lo que en la actualidad muchos autores lo denominan polineuromiopatía del paciente crítico (PNMPC)32,33.
Incidencia y etiopatogenia
La incidencia de la PPC es variable y oscila entre un 50 y un 80%, dependiendo fundamentalmente de los criterios diagnósticos utilizados34-36.
La etiología precisa de la PPC o la PNMPC continúa sin ser conocida. No obstante, en los últimos años se ha avanzado de forma considerable en el conocimiento de diversos factores asociados con el desarrollo de PPC. De Letter et al37, en un grupo heterogéneo de pacientes críticos, mostraron como factores independientes relacionados con la aparición de PPC la presencia de SRIS y la mayor gravedad de los enfermos. Nuestro grupo, en una cohorte de 73 pacientes críticos sépticos y con SDMO tras 10 días de VM, halló que la hiperosmolaridad, el empleo de nutrición parenteral, el uso de relajantes musculares y fallo neurológico (definido como GCS < 10) son factores de riesgo de PPC36. Por otro lado, un agente tóxico de bajo peso molecular no bien identificado ha sido detectado en sangre de estos pacientes y se ha propuesto que participa en el daño neuronal que sufren estos enfermos38.
Posteriormente, se han descrito en 95 pacientes críticos sometidos a VM durante más de 7 días como factores de riesgo independientes de neuropatía axonal aguda: el sexo femenino, el número de días con disfunción de dos o más órganos, la duración previa de la VM y la administración de corticoides39.
Un estudio prospectivo, aleatorizado y controlado diseñado para evaluar la acción del tratamiento insulínico intensivo con objeto de mantener una glucemia entre 80-110 mg/dl en paciente críticos quirúrgicos demostró una reducción significativa en la aparición de PPC en el grupo de tratamiento frente al grupo de tratamiento convencional (el 51,9 frente al 28,7%; p < 0,001)40. En un análisis multivariable se hallaron como factores independientes asociados con el desarrollo de PPC: tratamiento convencional con insulina, tratamiento con fármacos vasopresores por más de 3 días, bacteriemia y empleo de terapia de reemplazo renal. Esta disminución en la incidencia de PPC se observó incluso cuando se comparó el grupo de pacientes con glucemia entre 80-110 mg/dl con aquellos en que las cifras se mantuvieron en 100-150 mg/dl, y esta reducción estuvo en relación con las cifras de glucemia y no con la dosis de insulina aportada41. Posteriormente, en un ensayo clínico similar llevado a cabo por el mismo grupo en pacientes médicos, se demostró que la terapia intensiva insulínica es un factor protector del desarrollo de la PNMPC, lo que confirma nuestros hallazgos de que la administración de relajantes musculares es un factor de riesgo de PPC42.
Diagnóstico
Durante la fase aguda de la enfermedad crítica las manifestaciones de disfunción neuromuscular pueden enmascararse por la administración de sedantes y relajantes musculares, así como la encefalopatía que acompaña a muchos de estos procesos. Generalmente, la alteración a este nivel se pone de manifiesto cuando el paciente se recupera de la afección que lo condujo a la VM y se inicia la fase de desconexión del respirador43.
La forma de presentación es como tetraplejía o tetraparesia, a menudo con dificultad de desconexión del respirador44. Los reflejos osteotendinosos suelen estar abolidos, si bien podemos hallarlos reducidos o incluso normales. La concentración sérica de creatincinasa (CK) es normal o está ligeramente elevada. El LCR no presenta alteraciones patológicas.
Para el diagnóstico de la PPC es imprescindible un ENF que comprenda la realización de un electroneurograma y un electromiograma45. Aunque el momento de aparición de estos cambios no ha sido claramente determinado, se ha comprobado que una reducción patológica del potencial de respuesta motriz se detecta a los 2-5 días de inicio de la sepsis grave, asociándose precozmente con cambios miopáticos46. En el ENF45 se observa degeneración axonal primaria de fibras motrices y sensitivas con conservación de la velocidad de conducción, latencia distal normal y caída del potencial de respuesta motriz, que puede llegar a desaparecer en los casos graves si bien algunos autores han encontrado la afección predominantemente en las fibras motrices47. Es muy importante para el diagnóstico buscar denervación en los músculos como expresión de la agudeza del proceso, que se manifiesta por aparición en el EMG de potenciales de fibrilación y ondas positivas.
En la histología se observan cambios compatibles con una axonopatía que no afecta a la mielina ni produce signos inflamatorios48. No obstante, la biopsia nerviosa no es en absoluto necesaria para el diagnóstico de la PPC.
En la figura 1 se describe el algoritmo diagnóstico que se debe llevar a cabo ante la sospecha de enfermedad neuromuscular adquirida en el paciente crítico.
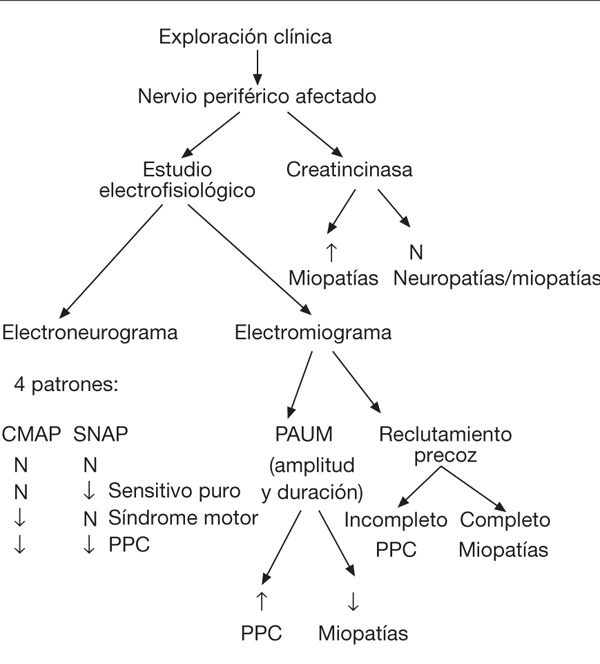
Figura 1. Protocolo diagnóstico ante sospecha de enfermedad neuromuscular adquirida en
el paciente crítico. CMAP: potencial de acción muscular compuesta; PAUM: potencial de acción
de unidad motriz; PPC: polineuropatía del paciente crítico; SNAP: potencial de acción sensitivo.
Tratamiento
Actualmente no existe tratamiento efectivo para la PPC. El tratamiento actual pasa por la identificación del problema e instauración de fisioterapia precoz. Por otro lado, la identificación de los factores de riesgo nos debe llevar a evitar el uso de relajantes musculares y a un estrecho control metabólico de los pacientes sépticos, evitando especialmente la hiperglucemia y la hiperosmolaridad. En el caso de que sea preciso el empleo de relajantes musculares, debemos emplear la menor dosis posible y en bolo, evitando la infusión continua para que así haya periodos sin relajación.
Pronóstico
En los casos de polineuropatía leve, la recuperación es favorable en semanas. En los casos de afección grave, el pronóstico funcional no es bueno, y a los 2 años persisten importante limitación de la movilidad y calidad de vida muy deteriorada en casi la totalidad de los pacientes evaluados49-51. La coexistencia de una neuropatía axonal con enlentecimiento de la velocidad de conducción se asocia a una peor recuperación. Una prolongada estancia en UCI, la mayor duración de la sepsis y la pérdida de peso son los tres parámetros que se asocian a una peor recuperación según un estudio reciente que siguió la evolución durante 2 años de 19 pacientes con PPC49.
Diversos estudios han evaluado la repercusión de la PPC sobre la retirada de la VM, con resultados inicialmente contradictorios36,52,53. Posteriormente, dos estudios prospectivos que incluyeron exclusivamente a pacientes recuperados de la enfermedad que los condujo a la VM, con buen nivel de conciencia y con criterios para comenzar la desconexión del respirador, estudiaron la influencia de la PPC en el retraso del destete, y observaron en el análisis multivariante que la PPC es un factor de riesgo independientemente asociado a prolongación de la desconexión de la VM54,55. En concreto, nuestro grupo en un estudio de 64 pacientes críticos sépticos encontró que el tiempo total de VM y la duración del periodo de desconexión del respirador fueron significativamente más prolongados en pacientes con PPC que en los que no tuvieron esta complicación. Igualmente, las reintubaciones y la necesidad de practicar traqueotomía fueron más frecuentes en el grupo con PPC56. Más recientemente, De Jonghe et al han demostrado la correlación entre la debilidad de las extremidades evaluada por una escala clínica y el descenso de los parámetros de función de los músculos respiratorios (presión inspiratoria máxima, presión espiratoria máxima y capacidad vital)57. Estos cambios se asocian a un aumento del tiempo de soporte respiratorio.
También ha habido controversia respecto a la influencia de la PPC o la PNMPC en la mortalidad. Así, Leijten et al53 encontraron más mortalidad en UCI entre los pacientes con PPC, aunque este dato podía explicarse por la mayor gravedad al ingreso de estos pacientes, más que por la contribución específica de la complicación neurológica. Esta diferencia de mortalidad no persistía tras 1 año de seguimiento. En contraste, De Jonghe et al39, en un estudio multicéntrico de pacientes críticos, no encontraron diferencias en la mortalidad de UCI entre ambos grupos, aunque la mortalidad total fue inesperadamente baja (8,4%). Nuestro grupo fue el primero en demostrar que la PPC es un factor independiente de mortalidad hospitalaria en pacientes con sepsis grave o shock séptico que requiere VM durante al menos 10 días36.
Trastornos de la conducción neuromuscular
El bloqueo neuromuscular prolongado tras el uso de pancuronio y vecuronio, especialmente cuando hay insuficiencia hepática o renal, se debe a la acumulación de los 3-hidroxi y 3-desacetil metabolitos que poseen actividad. Diversos trastornos metabólicos tales como hipofosfatemia y en especial la hipermagnesemia pueden exacerbar el bloqueo. El diagnóstico se confirma con la estimulación repetitiva a 3 y 20 Hz de los nervios periféricos que muestra una reducción progresiva de la transmisión nerviosa. La recuperación ocurre en días o semanas.
Miopatía del paciente crítico
En 1977, MacFarlane et al describieron por primera vez el desarrollo de una miopatía grave en una mujer joven tras el tratamiento de un status asmático con corticoides a elevadas dosis y relajantes musculares58.
Se han realizado diversas clasificaciones histológicas de la miopatía aguda en el paciente crítico. Recientemente, Hund propuso la clasificación de miopatía del paciente crítico (MPC), miopatía de filamento grueso y miopatía necrosante basada en los cambios histológicos presentes y que ha sido ampliamente aceptada59. No se conoce si estas tres formas representan diversos estadios de un mismo proceso, y es frecuente que en una biopsia muscular aparezcan en diversos grados estos tres patrones.
Desde el punto de vista práctico, podemos separar dos situaciones distintas: la MPC que afecta a enfermos con sepsis grave y SDMO, la cual se asocia generalmente a la PPC y la miopatía cuadripléjica aguda (MCA) de los pacientes asmáticos que reciben altas dosis de esteroides y relajantes musculares no despolarizantes (RMND). La MPC se trata de una miopatía primaria, es decir, no secundaria a la denervación muscular32.
Incidencia y etiopatogenia
La incidencia de MPC no es bien conocida. La principal razón es que la miopatía es difícil de detectar, ya que los ENF no son sensibles ni específicos y se requiere un procedimiento invasivo como es la biopsia muscular60-62. Recientemente, hemos realizado un estudio prospectivo en 26 pacientes ingresados por reagudización grave de EPOC que recibieron altas dosis de esteroides intravenosos. En el momento del destete, todos fueron estudiados con ENF. De ellos, 9 (34,6%) desarrollaron miopatía, que se confirmó por biopsia en los casos en que se realizó63.
Los mecanismos íntimos no son bien conocidos, y se han propuesto muy diversos factores que pueden contribuir al desarrollo de MPC. Por un lado, tendríamos la repercusión de la sepsis en el músculo y por otro, el papel de los corticoides y los RMND que debido al aumento de la permeabilidad vascular que hay en la sepsis fácilmente pueden acceder al músculo y dañarlo.
En el músculo existen cuatro sistemas proteolíticos: complejo ubiquitina-proteasoma, proteasas lisosomales, proteasas dependientes del calcio (calpaína) y proteasas no lisosomales que no dependen de calcio o ATP. Diversos mediadores proinflamatorios implicados en la sepsis (TNF α e IL-1 principalmente) inducen proteólisis. Estas citocinas activan la ubiquitina-proteasoma, principal vía intracelular de degradación proteica64. En contraposición, Showalter et al65 describieron la proteólisis mediada por la activación de la calpaína como factor etiopatogénico principal en pacientes con MPC.
Recientemente, se ha demostrado en un modelo animal de sepsis (ligadura y punción del ciego) que la debilidad muscular que presentaban las ratas se asociaba a la presencia de anticuerpos contra el receptor de la acetilcolina al igual que ocurre en la MG66. Este estudio puso de manifiesto la posibilidad de que un mecanismo inmunológico causara una disminución de los receptores nicotínicos en músculo esquelético, lo cual explicaría la debilidad muscular, si bien no se ha comunicado este hallazgo en humanos. Además, aunque la ausencia de infiltrado inflamatorio es casi constante en la microscopia, se encontró la presencia de marcadores inmunohistoquímicos de inflamación en pacientes con MPC, lo que indica que un mecanismo inflamatorio puede participar en la patogenia de esta entidad67.
Por otro lado, es bien conocido desde hace décadas que la administración crónica de corticoides causa miopatía. Tras la denervación experimental, se produce un incremento en el número de receptores de corticoides en el músculo estriado, lo que incrementa la sensibilidad del músculo a concentraciones normales de corticoides. Esto explicaría el efecto aditivo sobre el músculo de los RMND y los esteroides. Además, los corticoides aumentan el catabolismo de las proteínas musculares y disminuyen su síntesis. Se ha demostrado en un estudio en ratas que la administración de altas dosis de corticoides provoca debilidad por atrofia muscular dependiente de la dosis, y no por cambios en la expresión del receptor de acetilcolina que típicamente se observa tras el empleo de relajantes musculares68,69.
Manifestaciones clínicas y diagnóstico
El cuadro clínico de una miopatía es indistinguible de otras etiologías de enfermedad neuromuscular adquiridas en UCI (tabla 4). Estos pacientes presentan debilidad simétrica de las extremidades y los músculos respiratorios. Los reflejos profundos suelen estar reducidos o ausentes.

La CK sérica está dentro de la normalidad o ligeramente elevada, excepto en los casos de miopatías necrosantes, que generalmente tienen una marcada elevación. El diagnóstico se basa en el ENF y en la biopsia muscular. El EMG con aguja registra el patrón de actividad en el músculo tanto en reposo como en actividad. Los dos signos que claramente pueden diferenciar entre neuropatía y miopatía son el análisis del potencial de acción de unidad motriz (PAUM) y el reclutamiento de fibra generado por el esfuerzo voluntario. En las miopatías, el PAUM está disminuido en amplitud y duración y es polifásico, con reclutamiento precoz (fig. 2). En individuos sanos pueden recogerse PAUM polifásicos aislados, y se considera patológico cuando aparecen en más del 20% de los potenciales registrados. Obviamente, dado que para la obtención de PAUM se requiere la cooperación del paciente, puede haber una subestimación de la frecuencia de la miopatía en pacientes críticos45.

Figura 2. Registro de potenciales de acción de unidad motora (PAUM) obtenidos tras contracción
muscular voluntaria. En la imagen de la izquierda, observamos un patrón normal, a diferencia de
la imagen de la derecha donde se registran los PAUM de pequeño voltaje y polifásicos compatibles
con un patrón miopático.
El diagnóstico de certeza lo da la biopsia muscular, pero no hay que olvidar que es un método invasivo y puede no ser reproducido con facilidad. Actualmente se debería realizar la biopsia a los pacientes con debilidad muscular, especialmente si impide la desconexión de la VM, sin un diagnóstico neurofisiológico de PPC ni bloqueo de conducción en el ENF.
La microscopia óptica con tinción con hematoxilina-eosina muestra fibras anguladas atróficas, predominantemente del tipo II, con citoplasma basófilo. Las fibras atróficas se tiñen más débilmente con ATP-asa hacia el centro de la fibra, presentando una buena correlación con la microscopia electrónica. Esta última, además, puede revelar una pérdida generalizada de todo tipo de filamentos, pero lo más característico es la pérdida selectiva de las fibras gruesas de miosina, lo cual es casi patognomónico de la MPC32,48. En la miopatía necrosante hay mionecrosis extensa y fagocitosis de las fibras musculares61.
Tratamiento y pronóstico
No existe tratamiento específico. Aunque su papel en el pronóstico de los pacientes críticos no está bien definido debemos evitar aquellos agentes que propicien su aparición e iniciar una rehabilitación precoz de los pacientes afectos.
El impacto en el tiempo de VM o la mortalidad de la MPC no está bien dilucidado54. En un reciente estudio prospectivo de pacientes con EPOC, hemos demostrado que el desarrollo de MPC incrementa el tiempo de VM y estancia hospitalaria, pero no influye en la mortalidad63.
En el caso de la miopatía necrosante, la mortalidad es superior al 70%, aunque el papel de la miopatía en el fallecimiento no ha sido establecido61. De igual modo, se ha constatado un incremento de estancia hospitalaria en los pacientes que tras un trasplante hepático sufren una miopatía necrosante70.
En esta revisión hemos intentado resumir las principales enfermedades neuromusculares que pueden estar presentes en los pacientes críticos, tanto las previas al ingreso en UCI, que por su gravedad pueden requerir admisión en la unidad, como las adquiridas en UCI, las cuales se ponen de manifiesto en el momento de recuperación de la enfermedad y del inicio del destete de la VM. Es fundamental optimizar el manejo médico de estos pacientes, preferiblemente en una UCI especializada, con el objetivo de disminuir la aún elevada morbimortalidad asociada a estas afecciones. Se consideran necesarios futuros estudios para avanzar en el conocimiento de la patogenia de estas entidades, lo cual nos podría llevar a encontrar un tratamiento específico y efectivo.
Bibliografía
1. Mehta S. Neuromuscular disease causing acute respiratory failure. Respir Care. 2006;51:1016-21. [ Links ]
2. Van der Meché FG, Van Doorn PA, Meulstee J, Jennekens FG; GBS-consensus group of the Dutch Neuromuscular Research Support Centre. Diagnostic and classification criteria for the Guillain-Barré syndrome. Eur Neurol. 2001;45:133-9. [ Links ]
3. Hughes RA, Cornblath DR. Guillain-Barré Syndrome. Lancet. 2005;366:1653-66. [ Links ]
4. Guillain-Barré Syndrome Study Group. Guillain-Barré Syndrome: an Italian multicentre case-control study. Neurol Sci. 2000; 21:229-34. [ Links ]
5. Kiefer R, Kieseier BC, Hartung HP. Immune-mediated neuropathies. En: Pourmand R, Harati Y, editores. Neuromuscular disorders. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 111-31. [ Links ]
6. Hughes RA, Wijdicks EF, Benson E, Cornblath DR, Hahn AF, Meythaler JM, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62:1194-8. [ Links ]
7. Freeman R. Autonomic peripheral neuropathy. Neurol Clin. 2007;25:277-301. [ Links ]
8. Taguchi Y, Takashima S, Sasahara E. Inappropriate secretion of antidiuretic hormone in a patient with chronic inflammatory demyelinating polyneuropathy. Intern Med. 2005;44:65-7. [ Links ]
9. Papazian O, Alfonso I. Polirradiculoneuropatías autoinmunes agudas. Rev Neurol. 2002;34:169-77. [ Links ]
10. The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985;35:1096-104. [ Links ]
11. Mc Khann GM, Griffin JW, Cornblath, Mellits ED, Fisher RS, Quaskey SA. Plasmapheresis and the Guillain-Barré syndrome: analysis of prognostic factors and the effect of plasmapheresis. Ann Neurol. 1988;23:347-53. [ Links ]
12. The French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Appopriate number of plasma exchanges in Guillain-Barré syndrome. Ann Neurol. 1997;41:298-306. [ Links ]
13. Hughes RA, Swan AV, Raphaël JC, Annane D, Van Koningsveld R, Van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130:2245-57. [ Links ]
14. Van der Meché FGA, Schmitz PIM, Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;326:1123-9. [ Links ]
15. Hughes RA, Raphael JC, Swan AV, Van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2001;(2):CD002063. [ Links ]
16. Plasma Exchange/Sandoglobulin Guillain-Barré Trial Group. Randomized trial of plasma exchange, intravenous immunoglobulin, and combined treatmets in Guillain-Barré syndrome. Lancet. 1997;349:225-30. [ Links ]
17. Guillain-Barré Syndrome Steroid Trial Group. Double-blind study of intravenous methylprednisolone in Guillain-Barré syndrome. Lancet. 1993;341:586-90. [ Links ]
18. Wollinsky KH, Hülser PJ, Brinkmeier H, Aulkemeyer P, Bössenecker W, Huber-Hartmann KH, et al. CSF filtration is an effective treatmen of Guillain-Barré syndrome. A randomized clinical trial. Neurology. 2001;57:774-80. [ Links ]
19. Koningsveld RV, Steyerberg EW, Hughes RA, Swan AV, Van Doorn PA, Jacobs BC. A clinical prognostic scoring system for Guillain- Barré syndrome. Lancet Neurol. 2007;6:589-94. [ Links ]
20. Visser LH, Schmitz PIM, Meulstee J, Van Doorn PA, Van der Meché FGA. Prognostic factors of Guillain-Barré syndrome after intravenous immunoglobulin or plasma exchange. Neurology. 1999;53:598-604. [ Links ]
21. De la Cour CD, Jakobsen J. Residual neuropathy in long-term population-based follow-up of Guillain-Barré syndrome. Neurology. 2005;64:246-53. [ Links ]
22. Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797-802. [ Links ]
23. Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843-54. [ Links ]
24. Scherer K, Bedlack R, Simel D. Does this patient have myasthenia gravis? JAMA. 2005;293:1906-914. [ Links ]
25. Jani-Acsadi A, Lisak R. Myasthenic crisis: Guidelines for prevention and treatment. J Neurol Sci. 2007;261:127-33. [ Links ]
26. Schneider-Gold C, Gajdos P, Toyka KV, Hohlfeld RR. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev. 2005;(2):CD002828. [ Links ]
27. Berrouschot J, Baumann I, Kalischewski P, Sterker M, Schneider D. Therapy of myasthenic crisis. Crit Care Med. 1997;25:1228-35. [ Links ]
28. Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Ann Neurol. 1997;41:789-96. [ Links ]
29. Hart IK, Sathasivam S, Sharshar T. Immunosuppressive agents for myasthenia gravis. Cochrane Database Syst Rev. 2007; (4):CD005224. [ Links ]
30. Calhoun R, Ritter JH, Guthrie TJ, Pestronk A, Meyers BF, Patterson GA, et al. Results of transcervical thymectomy for myasthenia gravis in 100 consecutive patients. Ann Surg. 1999;230:555-61. [ Links ]
31. Bolton CF, Gilbert JJ, Hahn AF, Sibbald WJ. Polyneuropathy in critically ill patient. J Neurol Neurosurg Psych. 1984;47:1223-31. [ Links ]
32. Latronico N, Fenzi F, Recupero D, Guarneri B, Tomelleri G, Tonin P, et al. Critical illness myopathy and neuropathy. Lancet. 1996;347:1579-82. [ Links ]
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;13:1154-9. [ Links ]
34. Stevens R, Dowdy D, Michaels R, Mendez-Tellez P, Pronovost P, Needham D. Neuromuscular dysfunction acquired in critical illness: a systematic review. Intensive Care Med. 2007;33:1876-91. [ Links ]
35. Witt NJ, Zochodne DW, Bolton CF, Maison FG, Wells G, Young B, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest. 1991;99:176-84. [ Links ]
36. Garnacho-Montero J, Madrazo-Osuna J, García Garmendia JL, Ortiz Leyba C, Jiménez-Jiménez FJ, Barrero-Almodóvar AE, et al. Critical illness polyneuropathy: Risk factors and clinical consequences. A cohort study in septic patients. Intensive Care Med. 2001;27:1288-96. [ Links ]
37. De Letter MA, Schmitz PI, Visser LH, Verheul FAM, Schellens RLLA, Op de Coul DAW, et al. Risk factors for the development of polyneuropathy and myopathy in critically ill patients. Crit Care Med. 2001;29:2281-6. [ Links ]
38. Druschky A, Herkert M, Radespiel-Tröger M, Druschky K, Hund E, Becker CM, et al. Critical illness polyneuropathy: clinical findings and cell culture assay of neurotoxicity assessed by a prospective study. Intensive Care Med.2001;27:686-93. [ Links ]
39. De Jonghe B, Sharshar T, Lefaucheur JP, Authier FJ, Durand-Zaleski I, Boussarsar M, et al. Paresis acquired in the intensive care unit. A prospective multicenter study. JAMA. 2002;288:2859-67. [ Links ]
40. Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359-67. [ Links ]
41. Van den Berghe G, Wouters PJ, Bouillon R, Weekers F, Verwaest C, Schetz M, et al. Outcome benefit of intensive insulin therapy in the critically ill: Insulin dose versus glycemic control. Crit Care Med. 2003;31:359-66. [ Links ]
42. Hermans G, Wilmer A, Masserman W, Milants I, Weuters PJ, Bobbaers H, et al. Impact of intensive insulin therapy on neuro-muscular complications and ventilator dependency in the medical intensive care unit. Am J Respir CritCare Med. 2007;175:480-9. [ Links ]
43. Bolton C. Neuromuscular manifestations of critical illness. Muscle Nerve. 2005;32:140-63. [ Links ]
44. Mesejo A, Pérez-Sancho E, Moreno E. Clinical consequences of neuromuscular impairments in critically ill patients. Nutr Hosp. 2006;21:104-13. [ Links ]
45. Bolton CF. Electrophysiologic studies of critically ill patients. Muscle Nerve. 1987;10:129-35. [ Links ]
46. Khan J, Harrison TB, Rich MM, Moss M. Early development of critical illness myopathy and neuropathy in patients with severe sepsis. Neurology. 2006;67:1421-5. [ Links ]
47. Hund E, Genzwurker H, Böhrer H, Jakob H, Thiele R, Hacke W. Predominant involvement of the motor fibres in patients with critical illness polyneuropathy. Br J Anaesthesia. 1997;78:274-9. [ Links ]
48. Coakley JH, Nagendran K, Yarwood GD, Honavar M, Hinds CJ.. . Patterns of neurophysiological abnormalities in prolonged critical illness. Intensive Care Med. 1998;24:801-7. [ Links ]
49. De Seze M, Petit H, Wiart L, Cardinaud JP, Gaujard E, Joseph PA, et al. Critical illness polyneuropathy. A 2-year follow-up study in 19 severe cases. Eur Neurol. 2000;43:61-9. [ Links ]
50. Herridge MS, Chenung AM, Tansey CM, Matte-Martyn A, Díaz-Granados N, Al-Saidi F, et al. One-year outcomes in survivors of acute respiratory distress syndrome. N Engl J Med. 2003;348:683-93. [ Links ]
51. Fletcher SN, Kennedy DD, Ghosh IR, Misra VP, Kiff K, Coakley JH, et al. Persistent neuromuscular and neurophysiologic abnormalities in long-term survivors of prolonged critical illness. Crit Care Med. 2003;31:1012-106. [ Links ]
52. Leijten FSS, Harinck-De Weerd JE, Poortvliet DC, De Weerd AW. The role of polyneuropathy in motor convalescence after prolonged mechanical ventilation. JAMA. 1995;274:1221-5. [ Links ]
53. Leijten FSS, De Weerd AW, Poortvliet DC, De Ridder VA, Ulrich C, Harinck-De Weerd JE. Critical illness polyneuropathy in multiple organ dysfunction syndrome and weaning from the ventilator. Intensive Care Med. 1996;22:856 61. [ Links ]
54. De Jonghe B, Bastuji-Garin S, Sharshar T, Outin H, Brochard L. Does ICU-acquired paresis lengthen weaning from mechanical ventilation? Intensive Care Med. 2004;30:1117-21. [ Links ]
55. Garnacho-Montero J, Amaya-Villar R, García-Garmendía JL, Madrazo-Osuna J, Ortiz-Leyba C. Effect of critical illness polyneuropathy on the withdrawal from mechanical ventilation and the length of stay in septic patients. Crit Care Med. 2005;33:349-54. [ Links ]
56. Garnacho-Montero J, Amaya-Villar R, Ortiz-Leyba C. Critical illness polyneuropathy. Crit Care Med. 2005;33:1675. [ Links ]
57. De Jonghe B, Bastuji-Garin S, Durand MC, Malissin I, Rodrigues P, Cerf C, et al. Respiratory weakness is associated with limb weakness and delayed weaning in critical illness. Crit Care Med. 2007;35:2007-15. [ Links ]
58. MacFarlane IA, Rosenthal FD. Severe myopathy after status asthmaticus. Lancet. 1977;2:615. [ Links ]
59. Hund E. Myopathy in critically ill patients. Crit Care Med. 1999;27:2544-7. [ Links ]
60. Lacomis D, Petrella JT, Giuliani MJ. Causes of neuromuscular weakness in the intensive care unit: A study of ninety-two patients. Muscle Nerve. 1998;21:610-7. [ Links ]
61. Helliwell TR, Coakley JH, Wagemakers AJ, Griffiths RD, Campbell IT, Green CJ, et al. Necrotizing myopathy in critical ill patients. J Pathol. 1991;164:307-14. [ Links ]
62. Behbehani NA, Al-Mane F, D'yachkova Y, Pare P, FitzGerald JM. Myopathy following mechanical ventilation for acute severe asthma: the role of muscle relaxants and corticosteroids. Chest. 1999;115:1627-31. [ Links ]
63. Amaya-Villar R, Garnacho-Montero J, García-Garmendía JL, Madrazo-Osuna J, Garnacho-Montero MC, Luque R, et al. Myopathy in patients intubated for exacerbation of chronic obstructive pulmonary disease. Intensive Care Med. 2005;31:157-61. [ Links ]
64. Tiao G, Hobler S, Wang JJ, Meyer TA, Luchette FA, Fischer JE, et al. Sepsis is associated with increased mRNAs of ubiquitinproteasome proteolytic pathway in human skeletal muscle. J Clin Invest. 1997;99:163-8. [ Links ]
65. Showalter CJ, Engel AG. Acute quadriplegic myopathy: Analysis of myosin isoforms and evidence for calpain-mediated proteolysis. Muscle Nerve. 1997;20:316-22. [ Links ]
66. Tsukagoshi H, Morita T, Takahashi K, Kunimoto F, Goto F. Cecal ligation and puncture peritonitis model shows a decrease nicotinic acetylcholine receptor numbers in rat muscle: Immunopathologic mechanisms? Anesthesiology. 1999;91:448-60. [ Links ]
67. Bazzi P, Moggio M, Prelle A, Sciacco M, Messina S, Barberini S, et al. Critically ill patients: immunological evidence of inflammation in muscle biopsy. Clin Neuropath. 1999;18:23-30. [ Links ]
68. Leatherman JW, Fluegel WL, David WS, Davies SF, Iber C. Muscle weakness in mechanically ventilated patients with severe asthma. Am J Respir Crit Care Med. 1996;153:1686-90. [ Links ]
69. Adnet F, Dhissi G, Borron SW, Galinski M, Rayeh F, Cupa M, et al. Complication profiles of adult asthmatics requiring paralysis during mechanical ventilation. Intensive Care Med. 2001;27:1729-36. [ Links ]
70. Campellone JV, Lacomis D, Kramer DJ, Van Cott AC, Giuliani MJ. Acute myopathy after liver transplantation. Neurology. 1998;50:46-53. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dra. R. Amaya Vilar.
Servicio de Cuidados Críticos y Urgencias.
Unidad de Cuidados Intensivos. Hospitales Universitarios Virgen del Rocío.
Avda. Manuel Siurot s/n. 41013 Sevilla. España.
Correo electrónico: ramayav@wanadoo.es
Aceptado el 9-12-2008.

