










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Intensiva
versión impresa ISSN 0210-5691
Med. Intensiva vol.36 no.1 ene./feb. 2012
Mecanismos biológicos involucrados en la propagación del daño en el traumatismo encéfalo craneano
Biological mechanisms involved in the spread of traumatic brain damage
M. Rovegnoa,b, P.A. Sotob, J.C. Sáezb y R. von Bernhardic
aLaboratorio de Neurociencias y Departamento de Medicina Intensiva, Facultad de Medicina, Pontificia Universidad Católica de Chile, Chile
bDepartamento de Fisiología, Facultad de Ciencias Biológicas, Pontificia Universidad, Católica de Chile, Chile
cLaboratorio de Neurociencias, Departamento de Neurología, Facultad de Medicina, Pontificia Universidad Católica de Chile, Chile
Financiación: Proyecto PMD-10/09, Dirección de Investigación de la Facultad de Medicina-PUC (M.R.). Beca de apoyo a la realización de tesis doctoral 2010, AT-24100202 CONICYT (M.R.). Fondecyt 1070591 (J.C.S.); Fondecyt 1090353 (R.vB.).
Dirección para correspondencia
RESUMEN
El traumatismo encéfalo craneano (TEC) es un problema de salud de distribución mundial, y es especialmente prevalente en la población adulta joven. Es característica la presencia de uno o más focos de daño, que luego progresan hacia áreas inicialmente no lesionadas, mediante cascadas de respuesta inflamatoria, excitotoxicidad, condiciones de falla energética, y la participación de la glía amplificando la respuesta tisular al daño inicial. Esta progresión es, en teoría, susceptible de una intervención terapéutica. Sin embargo, hasta ahora todos los estudios con fármacos neuroprotectores han fracasado, no existiendo un tratamiento específico efectivo. Los resultados negativos se explican en parte por el empleo de una estrategia centrada solo en las neuronas, sin considerar otras células participantes, u otros mecanismos patogénicos. Para cambiar este panorama, es necesario re-enfocar el problema a través de una mejor comprensión de los mecanismos que determinan la progresión del daño. En esta revisión discutiremos los principales mecanismos biológicos involucrados en la progresión del daño tisular post-trauma. Se aborda la fisiopatología general de los tipos de traumatismos, mecanismos celulares del daño secundario incluyendo inflamación, apoptosis, tumefacción celular, excitoxicidad, y participación de la glía en la propagación del daño. Se destaca el papel de la glía en cada uno de los mecanismos celulares mencionados. Se incluyen algunas aproximaciones terapéuticas relacionadas con los mecanismos descritos. Se finaliza con un diagrama general que resume los principales aspectos discutidos.
Palabras clave: Apoptosis. Daño cerebral. Excitoxicidad. Neuroinflamación. Neuroglía. Traumatismo craneoencefálico.
ABSTRACT
Traumatic brain injury (TBI) is a worldwide health problem that is especially prevalent in young adults. It is characterized by one or more primary injury foci, with secondary spread to initially not compromised areas via cascades of inflammatory response, excitotoxicity, energy failure conditions, and amplification of the original tissue injury by glia. In theory, such progression of injury should be amenable to management. However, all neuroprotective drug trials have failed, and specific treatments remain lacking. These negative results can be explained by a neuron centered approach, excluding the participation of other cell types and pathogenic mechanisms. To change this situation, it is necessary to secure a better understanding of the biological mechanisms determining damage progression or spread. We discuss the biological mechanisms involved in the progression of post-trauma tissue damage, including the general physiopathology of TBI and cellular mechanisms of secondary damage such as inflammation, apoptosis, cell tumefaction, excitotoxicity, and the role of glia in damage propagation. We highlight the role of glia in each cellular mechanism discussed. Therapeutic approaches related to the described mechanisms have been included. The discussion is completed with a working model showing the convergence of the main topics.
Keywords: Apoptosis. Brain injury. Excitotoxicity. Neuroinflammation. Neuroglia. Traumatic brain injury.
Relevancia del problema
El traumatismo encéfalo craneano (TEC) es la principal causa de morbimortalidad en la población menor de 45 años. Cada año, 1,4 millones de personas sufren un TEC en los EE.UU.1, la mitad de los cuales son severos (escala de Glasgow < 8), y tienen una mortalidad de ˜35% al año. En Europa el panorama no es diferente, la incidencia estimada es de 235 casos por 100.000 habitantes/año con una mortalidad de ˜11%2. Además, el TEC es un factor de riesgo conocido para el desarrollo de enfermedades crónicas neurodegenerativas como Alzheimer y Parkinson. Así, por su alta incidencia en la población activa y su morbimortalidad asociada, el TEC representa un problema de salud pública importante. Sin embargo, no existen tratamientos específicos efectivos para prevenir el daño asociado3. El objetivo de esta revisión es discutir los principales mecanismos biológicos involucrados en la progresión del daño post-trauma. Una mejor compresión del problema es un primer paso en el desarrollo de estrategias terapéuticas eficaces. La tabla 1 muestra los trabajos experimentales comentados en esta revisión, que evalúan aproximaciones terapéuticas asociadas a los distintos mecanismos biológicos estudiados.
Tipos de traumatismo encéfalo craneano según su mecanismo de daño directo
Dependiendo de la biomecánica del trauma, los TEC se clasifican en lesión 1) focal y 2) difusa. En la lesión focal por contacto directo, se encuentran los hematomas epi- y subdurales, contusiones y hematomas intraparenquimatosos4. En cambio, en la lesión difusa por aceleración/desaceleración, predominan los fenómenos de daño axonal difuso y edema cerebral4.
En las primeras horas post-TEC, en el cerebro se observan islotes de daño celular en la lesión focal versus un compromiso extenso en las lesiones difusas. En ambas áreas de daño se encuentran neuronas y células no neuronales necrosadas, con focos de hemorragia, que dependiendo de su cuantía, pueden constituir hematomas intraparenquimatosos5. Además, se desarrollan grados variables de edema y de tumefacción celular. El daño axonal difuso se caracteriza por una distribución amplia y asimétrica de tumefacciones axónicas5.
Propagación del daño
Una vez establecido el daño tisular directo, tanto en las lesiones focales como difusas, se desarrolla un fenómeno complejo de progresión del daño que involucra factores locales y sistémicos que en conjunto se conocen como mecanismos de daño secundario (fig. 1).
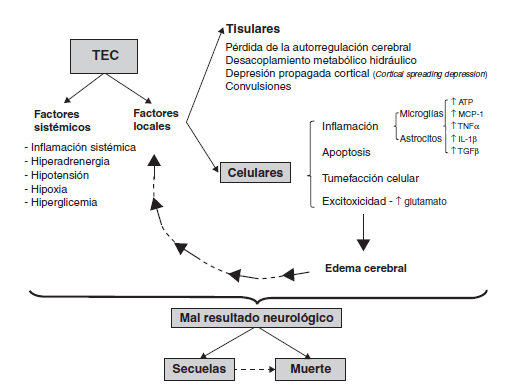
Figura 1. Mecanismos celulares y moleculares involucrados en el
traumatismo encéfalo craneano. Diagrama esquemático que
muestra la interrelación de factores sistémicos y locales en el
traumatismo encéfalo craneano. Entre los factores locales,
los factores celulares parecen ser especialmente importantes
en la amplificación y progresión del daño, independizándolo
de la noxa inicial. Para mayor información sobre los factores
tisulares y sistémicos ver cita bibliográfica4.
Entre los factores sistémicos que agravan el daño secundario destaca la presencia de hipotensión e hipoxia. El 90% de los pacientes que fallecen con un TEC grave presentan hallazgos compatibles con isquemia; hipotensión e hipoxia se observan en más del 30% de los pacientes y corresponden a dos de los cinco predictores de mortalidad más sólidos descritos para un TEC6.
Los factores locales de progresión del daño pueden ser subdivididos en tisulares y celulares. Los primeros comprenden, entre otros, aquellos dependientes de la relación existente entre la presión de perfusión cerebral y el flujo sanguíneo cerebral6. Posterior al daño tisular directo, se observa una caída del flujo sanguíneo cerebral, generándose una condición fisiopatológica similar a la isquemia4. Además, se producen lesiones extraaxiales (contusiones, hematomas, etc.), que inducen daño por compresión y herniación del parénquima cerebral. En forma más tardía se puede desarrollar hipertensión endocraneana (HEC), por aumento del volumen cerebral, debido a la presencia de lesiones extraaxiales y/o edema cerebral y a la naturaleza rígida de la bóveda craneana6. Los factores celulares corresponden a mecanismos patogénicos comunes a las neuronas y glías, como la presencia de inflamación, apoptosis, tumefacción celular y excitoxicidad. La glía, por sí misma, tiene un papel protagónico en la propagación del daño, coordinando y amplificando cada uno de los mecanismos patogénicos celulares recién mencionados. Los factores sistémicos y locales se potencian al interrelacionarse, lo cual determina por una parte, que la progresión del daño se independice de la noxa inicial (fig. 1), y por otra, que la presentación clínica sea heterogénea. Dado que el resultado final depende tanto del daño inicial como del secundario, y este a su vez de la interacción de los factores locales y sistémicos, son muchos los fenómenos patológicos en los cuales pueden existir diferencias caso a caso, amplificando la variabilidad. Esto ha dificultado enormemente la definición de dianas y el desarrollo de ensayos clínicos terapéuticos.
Una manera de enfrentar dicha heterogeneidad es diseñando estrategias «personalizadas», basadas en el genotipo del individuo. Al respecto, los estudios de asociación genética nos han demostrado que ciertos genes presentan polimorfismos que se correlacionan con el resultado clínico post-TEC7. En un metaanálisis reciente que incluyó a 2.527 pacientes, la isoforma ApoE4 de la apoproteína E se asoció a un mal resultado clínico a los seis meses post-TEC8. En cambio, un polimorfismo funcional del factor de supresión tumoral p53, se asoció a un mal pronóstico al momento del egreso de los pacientes con TEC admitidos a cuidados intensivos9. Los genes que codifican para las interleuquinas son también interesantes candidatos de estudio. La repetición de dos alelos del gen para el receptor antagonista de interleuquina 1 (IL-1ra) se relacionó con una presentación inicial más grave, pero con un mejor resultado clínico final10.
Inflamación
La respuesta inflamatoria post-TEC está mediada tanto por leucocitos circulantes, que son reclutados en la zona dañada, como por células residentes: microglías y astrocitos. Las microglías son macrófagos residentes del sistema nervioso central (SNC) y son responsables de la respuesta inmune innata del sistema nervioso11. Aun en ausencia de daño o estimulación exógena, las microglías despliegan una constante actividad de vigilancia remodelando continuamente sus procesos, pudiendo inspeccionar todo el parénquima cerebral en aproximadamente dos horas12. Al ser estimuladas, inician un proceso de activación donde exhiben respuestas de migración, fagocitosis, presentación de antígenos, producción de citoquinas, radicales libres, y otros factores solubles11. De esta forma, las microglías participan como sensores del daño, coordinadores y efectores de la respuesta inflamatoria del SNC.
Tras un TEC frecuentemente se produce la ruptura de la barrera hemato-encefálica, activación endotelial y reclutamiento e infiltración de leucocitos circulantes. Por su parte, las microglías se activan y migran al sitio de la lesión a través de varios mecanismos que incluyen quimioatractantes como el ATP liberado desde el foco de daño13, la proteína-1 quimioatractante de monocitos (MCP-1)14 y citoquinas como el factor de crecimiento transformante β (TGF-β)15. Tanto los leucocitos infiltrantes como las células residentes activadas liberan citoquinas pro-inflamatorias, óxido nítrico y radicales libres de oxígeno.
Los astrocitos son otro tipo de célula que participa en la respuesta neuroinflamatoria. Aquellos que sobreviven al daño celular inicial sufren un cambio fenotípico denominado astrocitosis reactiva, que se caracteriza por hipertrofia, hiperplasia y aumento de la expresión del filamento intermedio denominado proteína glial fibrilar acídica (GFAP). Este cambio fenotípico está mediado por el factor de necrosis tumoral α (TNF-α) y la interleucina 1-β (IL1-β)16. En un trauma moderado y focal, la astrocitosis contiene el área dañada y favorece la remodelación neuroplástica (arborización dendrítica y remodelamiento sináptico)17. Sin embargo, en TEC más severos, esta astrocitosis puede resultar en una cicatriz glial que posteriormente dificulta los cambios neuroplásticos necesarios para la acomodación funcional post-daño16.
Los diversos mediadores mencionados contribuyen a la perpetuación de la neuroinflamación. Sin embargo, esta respuesta neuroinflamatoria puede tener efectos neuroprotectores o neurotóxicos, dependiendo del tipo y magnitud del daño inicial y del contexto en el que ocurre la respuesta inflamatoria18.
La inflamación parece correlacionarse directamente con la magnitud del TEC y parece ser responsable al menos parcialmente del desarrollo de HEC. En una serie clínica de 23 pacientes con TEC grave, los niveles de TNF-α, IL-1β, IL-1α, IL-6 e IL-8 en el líquido céfalo-raquídeo (LCR) medidos por ELISA se elevaron precozmente en las primeras seis horas, alcanzando su máximo a las 12 horas y encontrándose niveles más elevados en los pacientes que desarrollaron HEC19.
Por otro lado, la producción de factores neurotróficos inducida por citoquinas, ejemplifica el potencial papel neuroprotector de la inflamación. En un estudio de 22 pacientes con TEC grave, tanto los niveles de IL-6 como del factor de crecimiento nervioso (NGF) se encontraron elevados en el LCR. La elevación de los niveles de NGF se detectó simultáneamente o a continuación de la aparición de la IL-6. Posteriormente, se observó que el LCR de pacientes con TEC estimuló la producción de NGF en cultivos de astrocito, lo que no se observó con el LCR de individuos controles sin TEC. Esta respuesta in vitro fue inhibida usando anticuerpos neutralizantes para la IL-620.
En el plano terapéutico, el uso de corticoides no ha resultado beneficioso para controlar la HEC a pesar de su potente efecto antiinflamatorio y de hecho, hay evidencias indicando que puede aumentar la mortalidad del TEC21. Sin embargo, aún no hay experiencias clínicas con aproximaciones antiinflamatorias más selectivas, como el uso de anticuerpos anticitoquinas, que podrían resultar eficaces. En un modelo murino de TEC focal, se evaluó la administración intratecal de un anticuerpo neutralizante de IL1-β. Su administración se inició cinco minutos post-trauma y se prolongó durante 14 días. Se observó que el anticuerpo neutralizante disminuyó la activación microglial, la infiltración por neutrófilos, la pérdida de tejido encefálico y los déficits neurológicos secundarios al traumatismo22.
Apoptosis
Durante un TEC se observan grados variables de daño sobre las células del parénquima cerebral. Los TEC severos resultan en una necrosis rápida, estableciéndose los focos de daño celular directo ya mencionados. Sin embargo, condiciones de daño sub-letales, o la propagación de señales de daño hacia sitios alejados del lugar de daño directo, determinan la aparición de muerte celular por apoptosis en dichas regiones23.
La existencia de apoptosis post-TEC ha sido observada en modelos animales y en pacientes24-26. En ratas, se observa apoptosis incluso posterior a un TEC leve24. La apoptosis presenta una distribución espacial y temporal particular, y es máxima en la sustancia gris y blanca adyacente al sitio de impacto cortical entre las 12 y 48 h post-trauma, disminuyendo de manera proporcional a la distancia desde la lesión, y es detectada hasta una semana post-lesión. Tardíamente, se observa la presencia de apoptosis alejada del trauma, en el tálamo ipsilateral a partir de las 72h post-TEC, con una evolución temporal retrasada respecto a la corteza correspondiente24.
Desde el punto de vista terapéutico, Clark et al.27 reportaron que la infusión intratecal de un tetrapéptido inhibidor de la caspasa-3 (enzima ejecutora de la apoptosis), en un modelo de TEC en ratas, disminuye la apoptosis y reduce significativamente la pérdida de tejido cerebral, evaluada a las tres semanas post-TEC. Sin embargo, los autores no encontraron diferencias en la evolución del compromiso neurológico27.
El inmunosupresor ciclosporina también reduce la pérdida de volumen cerebral post-TEC en varios modelos in vivo28,29. Actúa evitando la apertura del poro de permeabilidad transitoria mitocondrial, que ocurre por la sobrecarga de Ca2+ en la mitocondria observada durante episodios de isquemia-reperfusión y post-TEC30-32. Su análogo no inmunosupresor NIM811 fue equivalente a la ciclosporina en reducir la fragmentación del citoesqueleto y mejorar la respuesta motora en un modelo de TEC en ratones33. Considerando los efectos adversos derivados de su actividad inmunosupresora, NIM811 ofrece un mejor perfil de seguridad que la ciclosporina.
Tumefacción celular
El edema cerebral y las complicaciones asociadas dan cuenta de aproximadamente el 50% de las muertes por TEC34. Klatzo en 1967, usando un modelo de trauma por frío, describió dos tipos de edema cerebral dependiendo de su origen: edema vasogénico (extracelular) y citotóxico, el que más correctamente corresponde a una tumefacción celular35. Recientemente, mediante técnicas de neuroimagen, se determinó que el edema citotóxico es el componente mayoritario del edema cerebral post-TEC focal y difuso36.
El astrocito es el principal tipo celular que presenta tumefacción celular post-trauma37. Considerando que los astrocitos son diez veces más numerosos que las neuronas38, son el principal responsable del edema citotóxico. Aunque se han implicado numerosos factores, los mecanismos involucrados en la tumefacción son poco comprendidos. Se plantea que el daño celular directo y la hipoperfusión cerebral inicial ya mencionados producen consumo de ATP, falla energética y alteración de los gradientes iónicos transmembrana. En consecuencia, se produce despolarización neuronal y liberación de glutamato que induce una mayor entrada de Na+ y Ca2+ al interior de la célula. La sobrecarga de Na+ no alcanza a ser compensada por las bombas Na+/K+ ATPasa, debido en parte a la falla energética concomitante, favoreciendo la entrada de agua al interior celular, con el consiguiente aumento de volumen de la célula39.
Las acuaporinas (Acp) son canales de membranas y constituyen la vía principal de paso del agua a través de las membranas celulares. En el cerebro, la acuaporina principal es la Acp-4, y su distribución es altamente polarizada en las células ependimales y astrocitos40. Usando un modelo de edema cerebral citotóxico en un ratón knock out (KO), para Acp-4, se encontró una disminución significativa del edema y una mejoría en los parámetros neurológicos en los animales que carecen de la Acp-441. En el mismo ratón KO-Acp-4, también se observó una reducción en el contenido de agua cerebral y una mejor supervivencia, en un modelo de edema vasogénico por intoxicación acuosa41.
Dos trabajos recientes exploraron la manipulación farmacológica de la Acp-4. Usando Acp-4 reconstituida en liposomas, se demostró que el Zn2+ bloquea el paso de agua de manera rápida y reversible42. Por su parte, Kikuchi et al.43, utilizando edaravona (quelante de radicales libres) como tratamiento en un modelo de isquemia cerebral focal reversible, encontraron una disminución de la expresión de Acp-4 ipsilateral al infarto 24 horas post-reperfusión, que se acompañó de la disminución del área infartada y del déficit neurológico43.
Excitoxicidad
Tras un TEC, en un plazo de minutos, se observa una elevación aguda de los niveles extracelulares de glutamato, que ha sido documentada en trabajos experimentales y clínicos44,45. La elevación del glutamato extracelular es principalmente consecuencia de la despolarización neuronal masiva por el traumatismo y la falla energética asociada ya mencionada en la sección previa. Este exceso de glutamato induce un incremento de la entrada de Na+ y Ca2+ a la célula, la sobrecarga de Ca2+ intracelular desencadena mecanismos de daño celular que finalmente llevan al desarrollo de apoptosis por activación de caspasas46,47. La excitoxicidad por glutamato se produce inicialmente en las neuronas, en condiciones en las que los astrocitos representan la principal defensa al recapturar el glutamato. Los astrocitos captan el glutamato, por los transportadores de glutamato glutamato-1 (GLT-1) y el transportador de glutamato aspartato (GLAST), lo metabolizan a glutamina y lo tamponan espacialmente en conjunto con otros astrocitos acoplados a través de canales de uniones de hendidura (CUH)46. Los CUH están formados por la unión de dos hemicanales (HC), constituidos por hexámeros de conexinas (Cxs)48. Sin embargo, en una segunda etapa, si persisten concentraciones elevadas de glutamato extracelular, los astrocitos potencian la excitoxicidad ya que disminuyen los transportadores de glutamato49 y la sobrecarga de Na+ intracelular puede invertir el transporte50, favoreciendo la salida de glutamato desde el astrocito, aumentando aún más sus niveles extracelulares y promoviendo así el daño excitotóxico.
Dado que los efectos neurotóxicos del glutamato son conocidos desde hace más de 50 años51, inicialmente se pensó que la excitoxicidad podía ser la principal responsable de la fisiopatología del TEC. Esta visión ha sido replanteada debido a los numerosos resultados negativos en estudios terapéuticos en este campo52. El uso de antagonistas de los receptores de glutamato tipo N-metil-D-aspartato (NMDA), como selfotel o un bloqueador endógeno del mismo receptor, como el sulfato de magnesio, han fracasado en mostrar beneficios en estudios clínicos de pacientes con TEC53,54. Más aún, tanto el selfotel como el sulfato de magnesio aumentaron la mortalidad de los pacientes en comparación con el placebo53,54.
Propagación del daño, papel de la glía
Las glías, y los astrocitos en particular, considerados originalmente solo como soporte de las neuronas, han recibido poca atención en el estudio del TEC. Sin embargo, en la última década su papel ha sido reinterpretado. Los astrocitos forman la microarquitectura del sistema nervioso, expresan receptores, canales y estructuralmente están organizados en redes que se comunican a través de CUH, estableciendo sincitios funcionales55.
La progresión del daño celular podría estar facilitada por la comunicación intercelular mediada por canales formados por Cxs. En respuesta a la estimulación con diversos neurotransmisores o señales internas, los astrocitos pueden transmitir mensajes específicos a las células vecinas, principalmente a través de ondas de calcio, en lo que hoy se denomina gliotransmisión56, proceso en el que participan los CUH57. Además de los CUH, existen HC libres en regiones de la membrana que no establecen contactos celulares, los que comunican el interior de la célula con el medio extracelular. A través de los hemicanales, las células liberan moléculas biológicamente activas como glutamato, ATP y prostaglandinas58. De esta forma, se han demostrado dos vías de propagación para las ondas de calcio. Una vía intercelular mediada por CUH que permite el traspaso de moléculas como el inositol trifosfato (IP3), que inducen liberación de Ca2+ desde reservorios intracelulares y otra vía extracelular mediada por HC donde la principal molécula de señalización es el ATP48, que es liberada al medio extracelular y posteriormente señaliza en células distantes a través de los receptores purinérgicos P2.
Se ha propuesto que los CUH permiten la transferencia intercelular de señales de apoptosis59 mediando uno de los mecanismos de daño por vecindad, fenómeno que también se ha descrito en modelos de daño por transección de monocapas en cultivos celulares60. Las Cxs se denominan según sus pesos moleculares (en kilodaltons, ej.: Cx30, Cx43, Cx46)61; recientemente se demostró que los HC formados por Cx43 contribuyen a la propagación de la apoptosis en células alejadas del estímulo inicial62.
La modulación de la apertura de los HC se asocia al desarrollo de daño, observándose que un retardo en la apertura del HC inducida por inhibición metabólica se correlaciona con un retardo del daño celular63 y con una disminución en la liberación de ATP64. Entre los desencadenantes de la apertura de los HC se ha propuesto el aumento del potasio extracelular (K+e)65. Interesantemente, uno de los fenómenos característicos del trauma es la liberación del contenido intracelular, incluyendo el K+, al intersticio, como consecuencia del daño celular directo, alcanzándose concentraciones de K+e superiores a los 60 meq/L in vivo66. Este K+e, ya sea induciendo la apertura de canales de Ca2+67 o la apertura de los HC68, genera una sobrecarga de Ca2+ intracelular. La sobrecarga de Ca2+ intracelular puede inducir tanto necrosis como apoptosis por la activación de caspasas47.
Desde un punto de vista terapéutico, el bloqueo de los canales formados por conexinas (CUH y HC) podría tener un efecto beneficioso en el TEC. En un modelo de neurotrauma en cultivos organotípicos de hipocampo, se observó que el uso de bloqueadores de CUH, carbenoxolona y octanol, limitan el área de daño celular a solo el punto de impacto, impidiendo su progresión al resto de la rebanada. Además, la disminución de la Cx43 mediante el uso de RNAi también disminuyó la propagación de la muerte celular post-trauma23.
Daño axonal difuso
Una mención especial merece el daño axonal difuso, donde las lesiones comprometen fundamentalmente la sustancia blanca. La sustancia blanca está compuesta por haces axonales envainados con mielina, formada por los oligodendrocitos (OLG), encontrándose además astrocitos y células endoteliales. La interacción célula-célula entre los OLG y los axones neuronales es crítica en la formación de la mielina, el mantenimiento y la reparación de la sustancia blanca69.
El daño axonal difuso se presenta hasta en un 41% de los pacientes con TEC70, y es responsable de grados variables de compromiso de conciencia persistente. Clínicamente se caracteriza por una progresión rápida al coma sin que se observe una lesión intracraneana específica en las neuroimágenes. En la anatomía patológica se encuentran tumefacciones axónicas con una distribución difusa y asimétrica, que aparecen a las pocas horas post-trauma. Concomitantemente se observan focos de hemorragias a lo largo de los axones afectados y presencia de la proteína precursora de péptido β amiloide (Aβ). Posteriormente ocurre degeneración axonal, con desarrollo de los cuerpos de retracción5. Estos se producen cuando se interrumpe el transporte axonal por alteraciones del citoesqueleto, lo cual lleva al desarrollo de una tumefacción en el sitio de la desconexión que recibe el nombre de cuerpo de retracción71.
El daño axonal difuso tiene un resultado clínico distinto, desencadenándose una cascada de cambios que lleva a la desconexión secundaria de los axones. Según Povlishock, existen dos formas de daño axonal72. En la primera hay aumento de la permeabilidad axonal, tumefacción de las mitocondrias y degradación del citoesqueleto con compactación de los neurofilamentos. En la segunda existe una detención del transporte axonal y el desarrollo de tumefacción axonal, pero sin aumento de la permeabilidad axonal72. Lamentablemente, en el plano terapéutico, el daño axonal difuso es el área menos estudiada y hasta la fecha hay pocos estudios que nos permitan vislumbrar alguna aproximación con proyección terapéutica.
Recapitulación
Durante un TEC se establecen focos de daño celular directo. Sin embargo, diversos mecanismos de propagación de daño, tanto locales como sistémicos, independizan la progresión del daño de la noxa inicial. Entre los mecanismos locales destaca el papel de las glías, tanto de la microglía que participa como sensor del daño y coordinador de la respuesta inflamatoria, secretando numerosos mediadores de citotoxicidad, como de los astrocitos, los cuales, estando altamente interconectados, participan activamente en los procesos de inflamación, producción de radicales libres, tumefacción celular, excitoxicidad y propagación de la apoptosis. Los HC y CUH permitirían amplificar varias de estas respuestas, ya sea facilitando la propagación del daño o contribuyendo directamente al mismo. Los OLG, por su parte, tienen un papel preponderante en la homeostasis de la sustancia blanca, que es la principal afectada en el daño axonal difuso. La mayor parte de los estudios clínicos han sido diseñados con la intención de activar mecanismos neuroprotectores. Sin embargo, dados los resultados negativos obtenidos y considerando los mecanismos fisiopatológicos subyacentes a la propagación del daño post-TEC, parece ser necesaria una nueva aproximación que aborde los mecanismos de propagación de daño mediados por la glía.
Conflicto de interés
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006; 21:375-8. [ Links ]
2. Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien). 2006; 148:255-68. [ Links ]
3. Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002; 19:503-57. [ Links ]
4. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007; 99:4-9. [ Links ]
5. Adams JH, Graham DI, Scott G, Parker LS, Doyle D. Brain damage in fatal non-missile head injury. J Clin Pathol. 1980; 33:1132-45. [ Links ]
6. Chesnut RM. Care of central nervous system injuries. Surg Clin North Am. 2007; 87:119-56. [ Links ]
7. Dardiotis E, Fountas KN, Dardioti M, Xiromerisiou G, Kapsalaki E, Tasiou A, et al. Genetic association studies in patients with traumatic brain injury. Neurosurg Focus. 2010; 28:E9. [ Links ]
8. Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008; 25:279-90. [ Links ]
9. Martínez-Lucas P, Moreno-Cuesta J, García-Olmo DC, Sánchez-Sánchez F, Escribano-Martínez J, Pozo AC del, et al. Relationship between the Arg72Pro polymorphism of p53 and outcome for patients with traumatic brain injury. Intensive Care Med. 2005; 31:1168-73. [ Links ]
10. Hadjigeorgiou GM, Paterakis K, Dardiotis E, Dardioti M, Aggelakis K, Tasiou A, et al. IL-1RN and IL-1B gene polymorphisms and cerebral hemorrhagic events after traumatic brain injury. Neurology. 2005; 65:1077-82. [ Links ]
11. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009; 27:119-45. [ Links ]
12. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005; 308:1314-8. [ Links ]
13. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005; 8:752-8. [ Links ]
14. Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol. 2003; 54:388-92. [ Links ]
15. De Simone R, Ambrosini E, Carnevale D, Ajmone-Cat MA, Minghetti L. NGF promotes microglial migration through the activation of its high affinity receptor: modulation by TGF-beta. J Neuroimmunol. 2007; 190:53-60. [ Links ]
16. Laird MD, Vender JR, Dhandapani KM. Opposing roles for reactive astrocytes following traumatic brain injury. Neurosignals. 2008; 16:154-64. [ Links ]
17. Wieloch T, Nikolich K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol. 2006; 16:258-64. [ Links ]
18. Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007; 38:1392-400. [ Links ]
19. Hayakata T, Shiozaki T, Tasaki O, Ikegawa H, Inoue Y, Toshiyuki F, et al. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock. 2004; 22:102-7. [ Links ]
20. Kossmann T, Hans V, Imhof HG, Trentz O, Morganti-Kossmann MC. Interleukin-6 released in human cerebrospinal fluid following traumatic brain injury may trigger nerve growth factor production in astrocytes. Brain ResV 713. 1996; 143-52. [ Links ]
21. Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005; 365:1957-9. [ Links ]
22. Clausen F, Hånell A, Björk M, Hillered L, Mir AK, Gram H, et al. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009; 30:385-96. [ Links ]
23. Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neuroscience. 2002; 22:644-53. [ Links ]
24. Raghupathi R, Conti AC, Graham DI, Krajewski S, Reed JC, Grady MS, et al. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in cellular Bcl-2 immunoreactivity. Neuroscience. 2002; 110:605-16. [ Links ]
25. Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999; 13:813-21. [ Links ]
26. Miñambres E, Ballesteros MA, Mayorga M, Marin MJ, Muñoz P, Figols J, et al. Cerebral apoptosis in severe traumatic brain injury patients: an in vitro, in vivo, and postmortem study. J Neurotrauma. 2008; 25:581-91. [ Links ]
27. Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000; 74:740-53. [ Links ]
28. Scheff SW, Sullivan PG, Cyclosporin A. significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999; 16:783-92. [ Links ]
29. Sullivan PG, Rabchevsky AG, Hicks RR, Gibson TR, Fletcher-Turner A, Scheff SW. Dose-response curve and optimal dosing regimen of cyclosporin A after traumatic brain injury in rats. Neuroscience. 2000; 101:289-95. [ Links ]
30. Hansson MJ, Persson T, Friberg H, Keep MF, Rees A, Wieloch T, et al. Powerful cyclosporin inhibition of calcium-induced permeability transition in brain mitochondria. Brain Res. 2003; 960:99-111. [ Links ]
31. Uchino H, Elmér E, Uchino K, Li PA, He QP, Smith ML, et al. Amelioration by cyclosporin A of brain damage in transient forebrain ischemia in the rat. Brain Res. 1998; 812:216-26. [ Links ]
32. Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999; 160:226-34. [ Links ]
33. Mbye LH, Singh IN, Carrico KM, Saatman KE, Hall ED. Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J Cereb Blood Flow Metab. 2009; 29:87-97. [ Links ]
34. Miller JD, Becker DP, Ward JD, Sullivan HG, Adams WE, Rosner MJ. Significance of intracranial hypertension in severe head injury. J Neurosurg. 1977; 47:503-16. [ Links ]
35. Klatzo I. Presidental address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol. 1967; 26:1-14. [ Links ]
36. Marmarou A, Signoretti S, Fatouros PP, Portella G, Aygok GA, Bullock MR. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006; 104:720-30. [ Links ]
37. Barron KD, Dentinger MP, Kimelberg HK, Nelson LR, Bourke RS, Keegan S, et al. Ultrastructural features of a brain injury model in cat. I. Vascular and neuroglial changes and the prevention of astroglial swelling by a fluorenyl (aryloxy) alkanoic acid derivative (L-644,711). Acta Neuropathol. 1988; 75:295-307. [ Links ]
38. Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006; 29:547-53. [ Links ]
39. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004; 129:1021-9. [ Links ]
40. Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci USA. 1998; 95:11981-6. [ Links ]
41. Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000; 6:159-63. [ Links ]
42. Yukutake Y, Hirano Y, Suematsu M, Yasui M. Rapid and reversible inhibition of aquaporin-4 by zinc. Biochemistry. 2009; 48:12059-61. [ Links ]
43. Kikuchi K, Tancharoen S, Matsuda F, Biswas KK, Ito T, Morimoto Y, et al. Edaravone attenuates cerebral ischemic injury by suppressing aquaporin-4. Biochem Biophys Res Commun. 2009; 390:1121-5. [ Links ]
44. Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, DeKosky ST. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. 1993; 61:2015-24. [ Links ]
45. Bullock R, Zauner a , Woodward JJ, Myseros J, Choi SC, Ward JD, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998; 89:507-18. [ Links ]
46. Nishizawa Y. Glutamate release and neuronal damage in ischemia. Life Sci. 2001; 69:369-81. [ Links ]
47. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003; 4:552-65. [ Links ]
48. Bruzzone R, Giaume C. Connexins and information transfer through glia. Adv Exp Med Biol. 1999; 468:321-37. [ Links ]
49. Rao VL, Baskaya MK, Dogan A, Rothstein JD, Dempsey RJ. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem. 1998; 70:2020-7. [ Links ]
50. Allen NJ, Káradóttir R, Attwell D. Reversal or reduction of glutamate and GABA transport in CNS pathology and therapy. Pflügers Archiv. 2004; 449:132-42. [ Links ]
51. Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957; 58:193-201. [ Links ]
52. Jain KK. Neuroprotection in traumatic brain injury. Drug Discov Today. 2008; 13:1082-9. [ Links ]
53. Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg. 1999; 91:737-43. [ Links ]
54. Temkin NR, Anderson GD, Winn HR, Ellenbogen RG, Britz GW, Schuster J, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007; 6:29-38. [ Links ]
55. Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A. Glia: the fulcrum of brain diseases. Cell Death Differ. 2007; 14:1324-35. [ Links ]
56. Bezzi P, Volterra A. A neuron-glia signalling network in the active brain. Curr Opin Neurobiol. 2001; 11:387-94. [ Links ]
57. Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007; 161:61-79. [ Links ]
58. Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, et al. Connexin 43 hemichannels are permeable to ATP. J Neurosci. 2008; 28:4702-11. [ Links ]
59. Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, et al. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998; 1:494-500. [ Links ]
60. Cusato K, Bosco A, Rozental R, Guimarães CA, Reese BE, Linden R, et al. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003; 23:6413-22. [ Links ]
61. Sáez JC, Contreras JE, Bukauskas FF, Retamal MA, Bennett MVL. Gap junction hemichannels in astrocytes of the CNS. Acta Physiol Scand. 2003; 179:9-22. [ Links ]
62. Decrock E, De Vuyst E, Vinken M, Van Moorhem M, Vranckx K, Wang N, et al. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 2009; 16:151-63. [ Links ]
63. Contreras JE, Sánchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, et al. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002; 99:495-500. [ Links ]
64. Bahima L, Aleu J, Elias M, Martín-Satué M, Muhaisen A, Blasi J, et al. Endogenous hemichannels play a role in the release of ATP from Xenopus oocytes. J Cell Physiol. 2006; 206:95-102. [ Links ]
65. Srinivas M, Calderon DP, Kronengold J, Verselis VK. Regulation of connexin hemichannels by monovalent cations. J Gen Physiol. 2006; 127:67-75. [ Links ]
66. Nilsson P, Hillered L, Olsson Y, Sheardown MJ, Hansen AJ. Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J Cereb Blood Flow Metab. 1993; 13:183-92. [ Links ]
67. Duffy S, MacVicar BA. Potassium-dependent calcium influx in acutely isolated hippocampal astrocytes. Neuroscience. 1994; 61:51-61. [ Links ]
68. Sánchez HA, Orellana JA, Verselis VK, Sáez JC. Metabolic inhibition increases activity of connexin-32 hemichannels permeable to Ca2+ in transfected HeLa cells. Am J Physiol Cell Physiol. 2009; 297:C665-78. [ Links ]
69. Arai K, Lo EH. Oligovascular signaling in white matter stroke. Biol Pharm Bull. 2009; 32:1639-44. [ Links ]
70. Niess C, Grauel U, Toennes SW, Bratzke H. Incidence of axonal injury in human brain tissue. Acta Neuropathol. 2002; 104:79-84. [ Links ]
71. Smith DH, Meaney DF. Axonal Damage in Traumatic Brain Injury. Neuroscientist. 2000; 6:483-95. [ Links ]
72. Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. 2007; 161:43-59. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Correo electrónico: rvonb@med.puc.cl
(R. von Bernhardi)
Recibido el 1 de abril 2011
Aceptado el 25 de junio de 2011

