










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.32 no.3 Cantabria 2012
FGF23 and mineral metabolism, implications in CKD-MBD
Mariano Rodríguez1, Ignacio López2, Juan Muñoz1, Escolástico Aguilera-Tejero2, Yolanda Almaden1
1Servicio de Nefrología. Hospital Universitario Reina Sofía, REDINREN, IMIBIC. Córdoba
2Departamento de Medicina y Cirugía. Facultad de Veterinaria. Córdoba
The regulation of mineral metabolism is achieved trough a complex interaction of hormonal factors and target organs. Before the discovery of FGF23 we believed that the regulation of serum calcium and phosphate was mainly the result of changes in PTH and vitamin D acting on bone, kidneys and intestine. Parathyroids and kidneys were responsible for the production of PTH and 1,25(OH)2D3 respectively. Presently we know that FGF23 is produced by bone so the bone is not longer just a target organ but an active endocrine organ that participate in the regulation of mineral metabolism by sending signals through FGF23. Nephrologists are knowledgeable about the regulation of calcium and phosphate otherwise it is difficult to understand and manage the disturbances of mineral metabolism that are always present in patients with CKD. Changes in mineral metabolism in CKD are now described as chronic kidney disease-mineral and bone disorders (CKD-MBD).1 The pathology derived from CKD-MBD includes not only bone abnormalities but cardiovascular disease with a devastating prevalence of vascular calcification. The severity of CKD -MBD is associated with increased mortality in CKD patients.
The regulation of serum phosphate
The regulation of calcium and phosphate was only partially understood until the discovery of FGF23. FGF23 increases phosphaturia a reduces the production of 1,25(OH)2D3 (Figure 1). Let's think in a situation of hypocalcemia; the parathyroids respond promptly to a decrease in serum calcium, elevated PTH acts on bone to increase the exit of calcium, but the calcium release from bone is also accompanied by the release of phosphate. The PTH acts also in kidneys increasing the tubular reabsorption of calcium so the calcium released by bone is kept in the extracellular space. The PTH produces phosphaturia so the phosphate released by bone does not build up in the extracellular space. This may not be sufficient to bring the calcium up to normal, therefore the elevated PTH stimulatesrenal production of 1,25(OH)2D3 which in turn stimulates intestinal calcium absorption. This regulatory system appears to be adequate to control serum calcium, however 1,25(OH)2D3 not only increase gut absorption of calcium but also the absorption of phosphate. It does not seem logical that a synchronized hormonal response to correct hypocalcemia had to be concluded with an excess of phosphate. FGF23 modulates the production of 1,25(OH)2D3 and the accumulation phosphate. Both high phosphate and 1,25(OH)2D3 stimulate the production of FGF23 which feeds back on the production of1,25(OH)2D3 and induces phosphaturia. Thus the presence of FGF23 enables the system to restore the serum calcium without the trouble of phosphate accumulation (Figure 2).
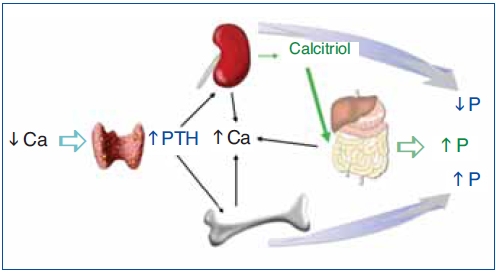
Figure 1. Hormonal response to hypocalcemia.
Ca: calcium; PTH: parathyroid hormone; P: phosphate.
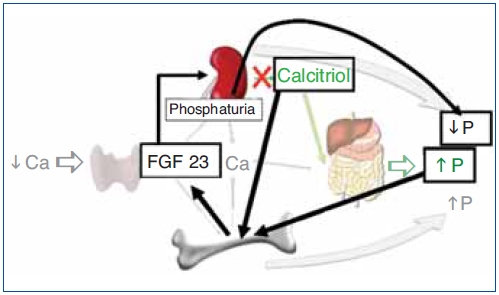
Figure 2. Hormonal response to hypocalcemia and the role of FGF23 to maintain phosphate balance.
Ca: calcium; FG23: fibroblast growth factor 23; P: phosphate; PTH: parathyroid hormone.
Production and actions of FGF23
FGF23 is a 32-kDa (251 amino acid) protein produced by osteocytes and osteoblasts which makes the bone an endocrine organ that communicates with other organs involved in mineral homeostasis. FGF23 acts on its receptor complex, klotho-FGFR1, in the kidney to cause phosphaturia and to decrease calcitriol synthesis.2-5 FGF23 induces phosphaturia by suppressing the expression ofthe Na-Pi cotransporters 2a and 2c in the brush border of renal proximal tubules. FGF23 suppresses renal production of 1,25(OH)2D3 by inhibiting 1α-hydroxylase (CYP27B1) activity which produces 1,25(OH)2D3 from 25(OH)D and also by increasing 24-hydroxylase activity which inactivates the 1,25(OH)2D3.3,6 Therefore the lack of FGF23, as in the FGF23 null mouse (FGF23-/-) causes hyperphosphatemia and high levels of 1,25(OH)2D3 a situation that produces extraosseous calcification.7 The endocrine action of FGF23 is dependent upon its binding and activation of the klotho-FGFR1 complex,5 therefore the absence of klotho as in the klotho-/-mouse produces a phenotype similar to the FGF23-/-mouse, elevation of phosphate and 1,25(OH)2D3 together with calcifications. We should be aware that these FGF23-/-rodents are teaching us what has been clinically evident in uremic patient: excessive doses of Calcitriol in combination with hyperphosphatemia carries the risk of calcification.
FGF23 production by osteoblasts and osteocytes is stimulated by high dietary intake of phosphate however the mechanisms at the cellular level are unknown.8 Experiments have failed to show a direct effect of high extracellular phosphate concentration on FGF23 expression by bone cells.8 The stimulation of FGF23 production by 1,25(OH)2D3 is well defined. Liu S et al.9 showed that 1,25(OH)2D3 up regulates FGF23 expression by acting on VDR response elements of the FGF23 promoter. Interestingly Carrillo et al.10 have shown thatestrogens directly stimulate the production of FGF23.
The interrelationship FGF23-PTH
Parathyroid tissue expresses a significant amount of klotho11 and FGF23 receptor. Thus it was reasonable to anticipate an effect of FGF23 on the parathyroids. FGF23 acts on the parathyroid FGF-Klotho complex12 causing activation ofthe MAPK pathway through ERK1/2 phosphorylation and increasein early growth response 1 mRNA levels. In vivo and in vitro experiments demonstrate that FGF23 decreased PTH mRNA and PTH secretion.12-14 FGF23 also produces upregulation of parathyroid 1 alpha hydroxilase expression.13 Canalejo et al.14 investigated the effect of FGF23 on two main parathyroid receptors that inhibit parathyroid function: the calcium sensing receptor and the vitamin D receptor. In vivo and in vitro studies demonstrated that FGF23 increased gene expression and protein levels of both calcium sensing and Vitamin D receptors. Finally the same authors showed that FGF23 decreased parathyroid cell proliferation. All these results strongly suggest that FGF23 inhibits parathyroid function in normal parathyroids. The expression of FGF23 receptor and klotho in parathyroids have been investigated. Some experiments have shown that administration of FGF23 produces upregulation of parathyroid klotho,13 other authors14 observed that FGF23 produced an increase in klotho that did not reach significance. High extracellular calcium was able to increase in both parathyroid klotho and FGF receptor expression in normal parathyroid glands.14
FGF23 in patients with chronic kidney disease. The pathogenesis of secondary hyperparathyroidism
Several publications have illustrated the important changes in FGF23 levels in patients with CKD.15-18 Some authors have shown that in early stages of CKD serum levels of FGF23 are elevated even when PTH is not significantly increased. For many years accumulation of phosphate and vitamin D deficiency were considered the key factors in the development of secondary hyperparathyroidism.18 The increase in serum PTH in CKD not only promotes urinary excretion of phosphate but also maintains serum calcium levels and stimulate the failing kidney to produce 1,25(OH)2D3. The increased production of FGF23 in CKD patients is most likely due to the increase in body burden of phosphate (not necessarily accompanied by hyperphosphatemia). FGF23 induces phosphaturia, which may explain why serum levels of phosphate are maintained in early stages of CKD. However FGF23 decreases de production of 1,25(OH)2D3 and accelerates its metabolism by augmenting 24(OH) asa activity. Thus, the decrease in 1,25(OH)2D3 seen in early CKD may be attributed not only to the decrease in renal mass but also to the early increase in FGF23.19 There is a debate about which of the two phosphaturic hormones, PTH or FGF23 increases earlier in CKD.20-22 An study by Isakova T et al.22 showed that in a group CKD patients with an average GFR of 41 ml/min had normal serum levels of calcium, phosphate and PTH, however FGF23 were already elevated and 1,25(OH)2D3 levels significantly reduced. Progressive loss of nephrons will make both FGF23 and PTH non-operative and then serum phosphate concentration will increase.
Nephrologists frequently ask whether or not it is advantageous to have elevation of FGF23. Certainly FGF23 helps to control phosphate balance but contributes to vitamin D deficiency. Furthermore recent experiments demonstrate a direct negative effect of FGF23 on the cardiovascular system.23 The fact that FGF23 is elevated indicates that the failing kidney needs the "help" of a phosphaturic hormone able to handle the phosphate load. Therefore the increase in FGF23 implies inadequate phosphate control. In patients with CKD a better control of phosphate is associated with a decrease in FGF23.24 Another question is whether FGF23 is a clinical usefultool to assess phosphate balance in CKD patients. FGF23 levels may not reflect acute changes in dietary phosphate; however high serum level of FGF23 may reveal a long period of positive phosphate balance. Certainly, clinical studies will have to be performed to prove the usefulness of FGF23 as a marker of phosphate balance.
A considerable amount of clinical studies have shown that a high FGF23 level is independent predictor of mortality,25-27 progression of renal disease28,29 left ventricular hypertrophy,30,31 vascular dysfunction,32 renal transplant outcome33 and experimental work have shown that FGF23 causes ventricular hypertrophy directly.23
FGF23 in advanced secondary hyperparathyroidism
In dialysis patients serum FGF23 levels are markedly increased and they are positively correlated with serum PTH levels and with serum levels of phosphate.16 One may assume that the sustained accumulation of phosphate is the cause of a direct correlation between PTH and FGF23. Nevertheless, given the fact that FGF23 inhibits parathyroid function it is unexpected to observe a parallel increase in the serum concentrations of FGF23 and PTH.
Experimental work in uremic rats with secondary hyperparathyroidism revealed that administration of FGF23 did not reduce serum levels of FGF23 in uremic rats; and, in vitro hyperplastic parathyroid glands from uremic rats did not respond to FGF23. Further experiments showed that hyperplastic parathyroid glands presented low expression of both FGF receptors and klotho. This results suggests a resistance of hyperplastic parathyroid gland to the inhibitory action of FGF23.14 Similar results were obtained by other group in another rat model of renal insufficiency.34 In parathyroid glands obtained from patients with advanced secondary hyperparathyroidism Klotho and FGFR1c expression decreased significantly particularly in glands with nodular hyperplasia.35-37
FGF23 after fenal transplant
After renal transplant many patients maintain high FGF23 levels suggesting that FGF23 may be the cause of of post-transplant hypophosphatemia with a relative vitamin D deficiency.38-40 Before transplantation FGF23 levels are very high and after kidney transplantation the excess of FGF23 acts to promote phosphaturia and suppress 1,25(OH)2D production. It is not clear why FGF23 secretion is maintained after transplantationdespite hypophosphatemia.
Conflict of interest
The authors declare potential conflicts of interest.
Grants: Amgen, Abbott, Fresenius
Presentation honoraria: Amgen, Abbott, Fresenius, Shire, Genzyme, Roche, Vifor
Consultant honoraria Amgen, Abbott, Fresenius, Shire, Genzyme, Roche, Vifor.
Referencias Bibliográficas
1. Moe S, Drüeke T, Cunningham J, Goodman W, Martin K, Olgaard K, et al.; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006;69(11):1945-53. [ Links ]
2. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 2001;98(11):6500-5. [ Links ]
3. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 2004;19:429-35. [ Links ]
4. Prie D, Urena TP, Friedlander G. Latest findings in phosphate homeostasis. Kidney Int 2009;75:882-9. [ Links ]
5. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006;444(7120):770-4. [ Links ]
6. Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol 2005;289:F1088-F1095. [ Links ]
7. Stubbs JR, Liu S, Tang W, Zhou J, Wang Y, Yao X, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol 2007;18:2116-24. [ Links ]
8. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 2005;90:1519-24. [ Links ]
9. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 2006;17:1305-15. [ Links ]
10. Carrillo-López N, Román-García P, Rodríguez-Rebollar A, Fernández-Martín JL, Naves-Díaz M, Cannata-Andía JB. Indirect regulation of PTH by estrogens may require FGF23. J Am Soc Nephrol 2009;20:2009-17. [ Links ]
11. Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, et al. Alpha-Klotho as a regulator of calcium homeostasis. Science 2007;316:1615-8. [ Links ]
12. Ben Dov IZ, Galitzer H, Lavi-Moshayoff, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest 2007;117:4003-8. [ Links ]
13. Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerström G, Jonsson KB, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 2007;195:125-31. [ Links ]
14. Canalejo R, Canalejo A, Martínez-Moreno JM, Rodríguez-Ortiz ME, Estepa JC, Mendoza FJ, et al. FGF23 fails to inhibit uremic parathyroid glands. J Am Soc Nephrol 2010;21:1125-35. [ Links ]
15. Larsson T, Nisbeth U, Ljunggren O, Jüppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 2003;64:2272-9. [ Links ]
16. Imanishi Y, Inaba M, Nakatsuka K, Okuno S, Yoshihara A, Miura M, et al. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int 2004;65:1943-6. [ Links ]
17. Shigematsu T, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, Fukagawa M. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis 2004;44:250-6. [ Links ]
18. Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. J Am Soc Nephrol 2011;6:913-21. [ Links ]
19. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 2010;78:975-80. [ Links ]
20. Isakova T, Wolf MS. FGF23 or PTH: which comes first in CKD? Kidney Int 2010;78:947-9. [ Links ]
21. Rodriguez M, Felsenfeld A. FGF 23, PTH in early renal failure. Nephrol Dial Transplant 2008;23(11):3391-4. [ Links ]
22. Isakova T, Gutierrez O, Shah A, Castaldo L, Holmes J, Lee H, et al. Postprandial mineral metabolism and secondary hyperparathyroidism in early CKD. J Am Soc Nephrol 2008;19(3):615-23. [ Links ]
23. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011;121(11):4393-408. [ Links ]
24. Gonzalez-Parra E, Gonzalez-Casaus ML, Galán A, Martinez-Calero A, Navas V, Rodriguez M, et al. Lanthanum carbonate reduces FGF23 in chronic kidney disease Stage 3 patients. Nephrol Dial Transplant 2011;26(8):2567-71. [ Links ]
25. Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008;359:584-92. [ Links ]
26. Jean G, Terrat JC, Vanel T, Hurot JM, Lorriaux C, Mayor B, et al. High levels of serum fibro- blast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant 2009;24:2792-6. [ Links ]
27. Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, et al.; Chronic Renal Insufficiency Cohort (CRIC) Study Group. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011;305(23):2432-9. [ Links ]
28. Fliser D, Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, et al.; MMKD Study Group. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 2007;18(9):2600-8. [ Links ]
29. Titan SM, Zatz R, Graciolli FG, Reis LM, Barros RT, Jorgetti V, et al. FGF-23 as predictor of renal outcome in diabetic nephropaty. Clin J Am Soc Nephrol 2011;6(2):241-7. [ Links ]
30. Hsu HJ, Wu MS. Fibroblast growth factor 23: a possible cause of left ventricular hypertrophy in hemodialysis pa tients. Am J Med Sci 2009;337:116-22. [ Links ]
31. Gutiérrez OM, Januzzi J, Isakova T, Laliberte K, Smith K, Collerone G, et al. Fibroblast growth factor-23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009;119:2545-52. [ Links ]
32. Mirza MA, Larsson A, Lind L, Larsson TE. Circulating fibro-blast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis 2009;205:385-90. [ Links ]
33. Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol 2011;22:956-66. [ Links ]
34. Galitzer H, Ben-Dov IZ, Silver J, Naveh-Many T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int 2010;77:211-8. [ Links ]
35. Komaba H, Goto S, Fujii H, Hamada Y, Kobayashi A, Shibuya K, et al. Depressed expression of Klotho and FGF receptor in hyperplastic parathyroid glands from uremic patients. Kidney Int 2010;77:232-8. [ Links ]
36. Kumata C, Mizobuchi M, Ogata H, Koiwa F, Nakazawa A, Kondo F, et al. Involvement of alpha-klotho and fibroblast growth factor receptor in the development of secondary hyperparathyroidism. Am J Nephrol 2010;31:230-8. [ Links ]
37. Krajisnik T, Olauson H, Mirza MA, Hellman P, Akerström G, Westin G, et al. Parathyroid Klotho and FGF-receptor 1 expression decline with renal function in hyperparathyroid patients with chronic kidney disease and kidney transplant recipients. Kidney Int 2010;78(10):1024-32. [ Links ]
38. Bhan I, Shah A, Holmes J, Isakova T, Gutierrez O, Burnett SM, et al. Post-transplant hypophos-phatemia: tertiary hyper-phosphatoninism? Kidney Int 2006;70:1486-94. [ Links ]
39. Evenepoel P, Naesens M, Claes K, Kuypers D, Vanrenterghem Y. Tertiary 'hyperphosphatoninism' accentuates hypophosphatemia and sup- presses calcitriol levels in renal transplant recipients. Am J Transplant 2007;7:1193-200. [ Links ]
40. Evenepoel P, Meijers BK, de Jonge H, Naesens M, Bammens B, Claes K, et al. Recovery of hyperphosphatoninism and renal phosphorus wasting one year after successful renal transplantation. Clin J Am Soc Nephrol 2008;3(6):1829-36. [ Links ]
![]() Correspondence:
Correspondence:
Mariano Rodríguez,
Servicio de Nefrología,
Hospital Universitario Reina Sofía,
REDINREN, IMIBIC, Córdoba
E-mail: juanm.rodriguez.sspa@juntadeandalucia.es
Enviado a Revisar: 25 Feb. 2012
Aceptado el: 25 Mar. 2012

