










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.32 no.5 Cantabria 2012
https://dx.doi.org/10.3265/Nefrologia.pre2012.Jun.11396
Biomarcadores en el síndrome nefrótico: algunos pasos más en el largo camino
Biological markers of nephrotic syndrome: a few steps forward in the long way
Alfonso Segarra-Medrano1, Clara Carnicer-Cáceres2, M. Antonia Arbós-Via3, M. Teresa Quiles-Pérez3, Irene Agraz-Pamplona1, Elena Ostos-Roldán1
1Servicio de Nefrología. Hospital Universitari Vall d'Hebron. Barcelona
2Servicio de Bioquímica. Hospital Universitari Vall d'Hebron. Barcelona
3Unidad de Investigación, Cirugía General. Hospital Universitari Vall d'Hebron. Barcelona
Dirección para correspondencia
RESUMEN
Uno de los retos a los que debe enfrentarse la nefrología moderna es el de identificar biomarcadores que se asocien a patrones anatomopatológicos o a mecanismos patogénicos definidos y permitan el diagnóstico no invasivo de la causa del síndrome nefrótico o establecer subgrupos pronósticos en cada tipo de enfermedad, prediciendo la respuesta al tratamiento y/o la aparición de recidivas. Los avances en el conocimiento de la patogenia de las distintas enfermedades causantes de síndrome nefrótico, sumados al progresivo desarrollo y estandarización de las técnicas de proteómica plasmática y urinaria, han permitido ir identificando un número creciente de moléculas que podrían ser útiles para los fines anteriormente mencionados. En el momento actual, los datos de muchos de los candidatos identificados, sobre todo mediante técnicas de proteómica, son todavía muy preliminares. En la presente revisión, se resume la evidencia disponible sobre las moléculas que en la actualidad cuentan con mayor evaluación en estudios clínicos.
Palabras clave: Biomarcadores, Síndrome nefrótico, CD80 urinario, Hemopexina, Receptor soluble IL-2, Receptor soluble uroquinasa, Anticuerpos anti receptor tipo M de la fosfolipasa, Beta-2 microglobulina, N-acetil-glucosaminidasa, Interleuquina 13.
ABSTRACT
One of the major challenges modern nephrology should face is the identification of biomarkers that are associated with histopathological patterns or defined pathogenic mechanisms that might aid in the non-invasive diagnosis of the causes of nephrotic syndrome, or in establishing prognosis sub-groups based on each type of disease, thus predicting response to treatment and/or recurrence. Advancements in the understanding of the pathogenesis of the different diseases that cause nephrotic syndrome, along with the progressive development and standardisation of plasma and urine proteomics techniques, have facilitated the identification of a growing number of molecules that might be useful for these objectives. Currently, the available information for many of the possible candidates identified to date, above all those discovered using proteomics, are still very preliminary. In this review, we summarise the available evidence for the different molecules that have been best assessed using clinical studies.
Key words: Biomarkers, Nephrotic syndrome, Urinary CD80, Hemopexyn, Soluble IL-2 receptor, Urokinase soluble receptor, Anti PLRA2 antibodies, Beta 2 microglobulin, N-acetyl-glucosaminidase, Interleukin 13.
Introducción
El síndrome nefrótico se define por la presencia de proteinuria superior a 3,5 g/día en adultos y 40 mg/m2 en niños, asociada a hipoalbuminemia, edemas, hiperlipidemia e hipercoagulabilidad1. El mecanismo común a todas las enfermedades renales causantes de este síndrome es la pérdida de la selectividad de la barrera de filtración glomerular, lo que permite el paso masivo de proteínas al espacio urinario2. Las formas primarias se definen como aquellas en las que no es posible hallar una enfermedad sistémica responsable de ello. Las formas secundarias incluyen las lesiones renales que aparecen como consecuencia de otras enfermedades que presentan, habitualmente, signos y síntomas extrarrenales. Las lesiones anatomopatológicas que con mayor frecuencia son responsables de síndrome nefrótico son la nefropatía por cambios mínimos (NCM), la glomeruloesclerosis focal y segmentaria (GFS), la nefropatía membranosa (NM) y, con menor frecuencia, la glomerulonefritis membranoproliferativa (GMP) entre las glomerulopatías primarias, y la nefropatía diabética y las nefropatías por depósito de inmunoglobulinas, entre las secundarias1-3. En los niños -por el marcado predominio de la NCM4,5- y en algunas formas secundarias del adulto, es posible tener un cierto grado de sospecha clínica sobre cuál es la lesión anatomopatológica causante del síndrome nefrótico. Sin embargo, en el momento actual, en la inmensa mayoría de los casos de síndrome nefrótico del adulto, es necesario realizar una biopsia renal para llegar a un diagnóstico de certeza, establecer un pronóstico y elegir el tratamiento más adecuado. Sin duda, uno de los retos pendientes a los que debe enfrentarse la nefrología moderna es el de identificar biomarcadores que se asocien a patrones anatomopatológicos o a mecanismos patogénicos definidos y que permitan el diagnóstico no invasivo de la causa del síndrome nefrótico o establecer subgrupos pronósticos en cada tipo de enfermedad, prediciendo la respuesta al tratamiento y/o la aparición de recidivas.
Los continuos avances en el conocimiento de la patogenia de las distintas enfermedades causantes de síndrome nefrótico, sumados al progresivo desarrollo y estandarización de las técnicas de proteómica plasmática y urinaria, han permitido ir identificando un número creciente de moléculas que podrían ser útiles para los fines anteriormente mencionados si se demuestra que tienen sensibilidad y especificidad suficiente para identificar el tipo de lesión renal y/o relación con la respuesta al tratamiento o con el pronóstico de la enfermedad. En el momento actual, los datos de muchos de los candidatos identificados -sobre todo mediante técnicas de proteómica- son todavía muy preliminares. En la presente revisión, se resume la evidencia disponible sobre las moléculas que en la actualidad cuentan con mayor evaluación en estudios clínicos.
Biomarcadores en la nefropatía por cambios mínimos
La NCM se caracteriza por la ausencia de lesiones visibles con técnicas de microscopía óptica y por la ausencia de depósitos en los estudios de inmunofluorescencia1,2. La única lesión demostrable es la fusión de los pies podocitarios en la microscopía electrónica. Las evidencias de una asociación frecuente con atopia, infecciones, vacunaciones y procesos linfoproliferativos, y de que la mayor parte de los enfermos responde al tratamiento con esteroides, inmunosupresores e inmunomoduladores, han sido argumentos sólidos para sugerir la participación del sistema inmunológico en su patogenia. En 1974, Shalhoub sugirió que la lesión de la barrera de filtración podía ser debida a la producción de una linfocina producida por los linfocitos T6. Desde entonces, numerosos estudios han demostrado la existencia de disregulaciones de la respuesta inmunitaria, especialmente en las células T, y se ha sugerido que la NCM podría ser la consecuencia de una alteración primaria en la función de estas células7-11. Se ha identificado un patrón de respuesta predominantemente Th2 durante la fase de actividad de la enfermedad12. Koyama et al.13 desarrollaron un hidridoma de células T capaz de inducir proteinuria a través de una modificación en la carga eléctrica de la barrera de filtración, y otros autores han aislado proteínas de origen monocitario y proteínas solubles relacionadas con la activación de la respuesta inmune, producidas por linfocitos T supresores, capaces de inducir proteinuria sin alterar la carga eléctrica de la barrera de filtración. La reciente evidencia de respuesta a rituximab en enfermos con dependencia de esteroides y anticalcineurínicos sugiere que los linfocitos B podrían desempeñar, de forma directa o en cooperación con los linfocitos T, un papel relevante en algunos enfermos14. A pesar de las claras evidencias de la implicación del sistema inmunológico en la patogenia de la NCM, todavía no se han identificado ni los mediadores ni los mecanismos de lesión podocitaria. Sin embargo, estudios realizados en los últimos años han aportado novedades relevantes sobre moléculas con posibilidad de ser utilizadas como biomarcadores relacionados con el diagnóstico, la monitorización de la actividad o la respuesta al tratamiento (figura 1 y tabla 1).
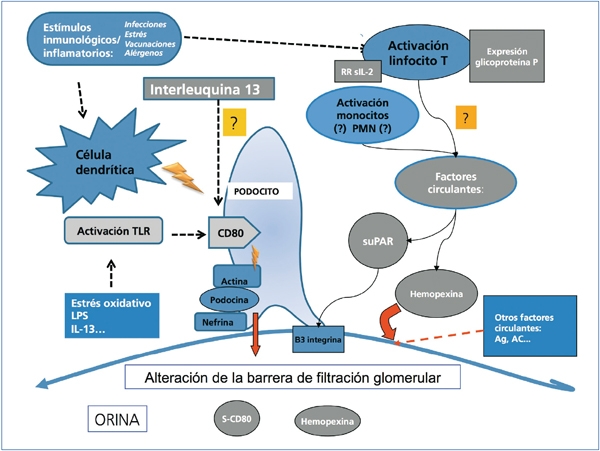
Figura 1. Esquema de los mecanismos patogénicos propuestos para explicar la lesión del podocito
en la nefropatía por cambios mínimos y en la glomeruloesclerosis focal y segmentaria de los posibles
biomarcadores que se encuentran en fase de estudio
AC: anticuerpos, Ag: Antigeno; IL-13: inlterleuquina 13; LPS: lipopolisacárido; PMN: polimorfonucleares;
RR sIL-2: forma soluble del receptor de membrana de interleuquina 2; suPAR: receptor soluble de uroquinasa;
TLR: receptores toll like.
Tabla 1. Listado de las características y potencial utilidad de los principales biomarcadores
propuestos para el estudio del síndrome nefrótico 
AR: aldosa-reductasa; GFS: glomeruloesclerosis focal y segmentaria; IF: inmunofluorescencia;
IL-13: interleuquina 13; NCM: nefropatía por cambios mínimos; NAG: N-acetil-glucosaminidasa;
NM: nefropatía membranosa; PCR: reacción en cadena de polimerasa; PLA2R: receptor tipo
M de la fosfolipasa A2; RR sIL-2: forma soluble del receptor de membrana de interleuquina 2;
SOD2: superóxido-dismutasa 2; suPAR: receptor soluble de uroquinasa.
Niveles urinarios y expresión podocitaria de CD80 (B7.1)
Datos muy recientes indican que las células podocitarias, en determinadas circunstancias, pueden adquirir fenotipo y/o funciones de células dendríticas y pueden ser inducidas a expresar CD80 (B7.1)15,16. CD80 es una proteína transmembrana expresada en células con capacidad presentadora de antígeno que, tras unirse a su ligando -CD28-, presente en los linfocitos T, proporciona una señal de coestimulación a éstos, imprescindible para la activación linfocitaria18. Los podocitos normales no expresan CD80. Se ha demostrado que, en modelos experimentales, la expresión de CD80 en podocitos se asocia a aparición de proteinuria nefrótica16. En la actualidad se ignora cuál es el significado funcional de la neoexpresión de CD80 por parte del podocito y no se ha podido demostrar su posible relación con las alteraciones de la membrana de filtración que causan proteinuria. La expresión de CD80 puede ser inducida por estrés oxidativo o tras estimulación con lipopolisacárido (LPS)16, a través de señalización mediada por receptores Toll like-318 e interleuquina (IL) 1319, y no es dependiente de linfocito, ya que puede demostrarse incluso en animales knock-out que carecen de linfocitos T16. La expresión de CD80 en podocitos ha sido demostrada en biopsias de NCM en humanos y recientemente se ha descrito que el nivel urinario de CD80 se halla elevado en los enfermos con NCM durante el brote y se normaliza tras la remisión, pero no se eleva en otras nefropatías causantes de síndrome nefrótico, como la NM o la GFS, ni en otros tipos de enfermedad glomerular20. Los datos clínicos disponibles son todavía preliminares, pero parecen indicar que la medición de CD80 urinario podría tener utilidad tanto para el diagnóstico no invasivo de NCM como para el diagnóstico diferencial entre NCM y GFS y/o para monitorizar la actividad de la enfermedad.
Interleuquina 13
A partir de la asociación entre NCM y enfermedad de Hodgkin, y de la evidencia de que IL-13 es un factor de crecimiento autocrino para las células de Reed-Sternberg22-24, se ha generado una amplia evidencia experimental -y en mucho menor grado clínica- que relaciona IL-13 con la inducción de alteraciones estructurales en el podocito, capaces de alterar la selectividad de filtración y de causar síndrome nefrótico. Recientemente, se ha demostrado que la expresión del gen IL-13 está aumentada tanto en linfocitos CD4 como en CD8 en niños con síndrome nefrótico corticosensible durante las recidivas12. Este aumento se asocia a niveles elevados de IL-13 en el citoplasma de las células T25 y a una infrarregulación en la expresión génica de citoquinas proinflamatorias IL-8 e IL-12 en monocitos26. Se ha descrito asimismo una correlación entre polimorfismos en la región 3 no traducida del gen de la IL-13 y la evolución clínica de la NCM. La expresión de ácido ribonucleico mensajero (mRNA) de IL-13 en células mononucleares en pacientes con haplotipo AAT -asociado a recaídas múltiples- es significativamente mayor que la observada en enfermos con haplotipo GCC -asociado a remisión a largo plazo27-. También se ha demostrado recientemente la presencia de receptores de IL-13 en podocitos y se ha descrito que la estimulación de podocitos en cultivo con IL-13 induce cambios funcionales consistentes en una disminución en la resistencia eléctrica transepitelial y una fosforilación de STAT628.
Se ha desarrollado también un modelo de ratón transgénico para IL-1329. En dicho modelo, los ratones transgénicos hiperexpresan y tienen niveles circulantes permanentemente elevados de IL-13, pero no de otras citoquinas Th1 (IL-2 o interferón [IFN]) o Th2 (IL-4) y desarrollan síndrome nefrótico con lesiones ultraestructurales renales idénticas a la NCM. Tanto los estudios de expresión génica como la inmunofluorescencia demostraron una reducción en la expresión de nefrina, podocina y distroglicanos y un incremento en la expresión podocitaria de CD80. En estudios experimentales previos, ya se había demostrado que IL-13, en combinación con IL-1 e IFN-gamma, podía inducir expresión de CD80 en células epiteliales tubulares proximales, pero no en podocitos. La evidencia de que IL-13 puede inducir expresión de CD80 en podocitos y que ésta se asocia a la aparición de síndrome nefrótico establece un vínculo de gran interés, ya que abre la posibilidad de considerar que en algún grupo de enfermos con NCM la proteinuria podría ser causada por efectos directos de IL-13 en la estructura terciaria del podocito y, a la vez, genera la hipótesis sobre la posible utilidad clínica del estudio de la vía IL-13-CD80 en enfermos con NCM, en relación con el curso clínico, respuesta al tratamiento y pronóstico. Por otra parte, la evidencia de que IL-13 tiene un papel clave en la producción de IgE e IgG4 en pacientes nefróticos, a diferencia de lo que ocurre en los pacientes con asma, en los que la producción de IgE es mayoritariamente dependiente de IL-430, podría contribuir a explicar la relación entre NCM y atopia.
Nivel sérico y actividad proteasa de la hemopexina circulante
La hemopexina plasmática (Hx) es una β-1 glicoproteína cuyo peso molecular es variable dependiendo del grado de glicosilación31,32. Además de su función básica, que consiste en unión y transporte del hemo libre y la homeostasis del hierro, Hx tiene actividad antioxidante33. Hx se considera como un reactante de fase aguda, ya que su síntesis hepática aumenta en respuesta a IL-6 e IL-134,35. Hay varias isoformas de Hx circulante que, hasta la fecha, han sido poco caracterizadas. Se considera que en sujetos sanos Hx circula en plasma en una forma inactiva. A partir de plasma humano normal, se ha identificado una isoforma con actividad proteasa que, in vitro, puede ser inhibida mediante varios inhibidores de la serina proteasa o mediante ATP36,37. Esta isoforma es capaz de inducir lesiones glomerulares similares a las observadas en la NCM tanto en tejido renal in vitro como tras infusión intrarrenal en ratas in vivo. La inducción de proteinuria y el borramiento podocitario se asocia a una reducción en la expresión de ecto-apirasa y retracción de los podocitos38,39.
La medición de los niveles séricos y urinarios y de la actividad proteasa de Hx indica que los enfermos con NCM en fase de actividad presentan unos niveles reducidos de Hx circulante y un incremento en su actividad proteasa. En la orina, a diferencia de lo que ocurre en otras enfermedades causantes de síndrome nefrótico, en los enfermos con NCM en fase de brote, la banda de Hx de 80 KD es prácticamente indetectable40. En conjunto, estos datos sugieren que en los enfermos con NCM, durante el brote, circula una isoforma de Hx con actividad proteasa incrementada, pero no se conoce cuál es el significado clínico de este dato. Son necesarios más estudios para determinar si estos datos son específicos de la NCM y si aportan algún valor en el diagnóstico o seguimiento de los enfermos.
Nivel sérico del receptor soluble de la interleuquina 2
El receptor de membrana de la IL-2 es una proteína formada por tres cadenas: alfa, beta y gamma41. El linfocito T no estimulado expresa las subunidades beta y gamma del receptor. Tras la activación del linfocito T subsecuente al reconocimiento de un antígeno a través del receptor de células T (TCR), asociada a señales de coestimulación, se expresa la cadena alfa, que juntamente a las otras dos formará el receptor de membrana funcionalmente activo de la IL-2 (IL-2R). En respuesta a la estimulación del TCR, el linfocito T sintetiza IL-2 que, mediante su unión al receptor de membrana (IL-2R), causará la activación y expansión clonal de los linfocitos T. Por razones poco conocidas, de forma paralela y proporcional a la expresión del IL-2R de membrana, se libera a la circulación una forma soluble del receptor (RR sIL-2) que se genera a partir de la ruptura proteolítica de la subunidad alfa. La función del receptor soluble no se conoce con certeza. Se considera que podría ser capaz de captar IL-2 circulante y, de esta manera, modular la cantidad de esta citoquina que se puede unir al receptor celular. El nivel circulante de RR sIL-2 se considera una medida indirecta de activación de células T42,43. Hay un gran número de evidencias44-52 que indican que durante la fase aguda los enfermos con NCM presentan niveles muy elevados de RR sIL-2 y que éstos se normalizan tras la remisión. Sin embargo, también se han descrito niveles elevados de RR sIL-2 en múltiples enfermedades inflamatorias e inmunológicas, en otras nefropatías primarias causantes de síndrome nefrótico y en la nefropatía lúpica y, hasta la fecha, no se ha realizado ningún estudio para analizar su sensibilidad y su especificidad, por lo que no es posible conocer si la determinación de los niveles circulantes del RR sIL-2 aporta algún valor diagnóstico o pronóstico en relación con los estudios convencionales.
ABCB1 y glicoproteína-P
La glicoproteína-P (CD243) es una proteína transmembrana, miembro de la familia de los ATP-binding cassette transporters (ABC-transporters), cuya síntesis está codificada por el gen ABCB1 (anteriormente denominado MDR1), localizado en la región 7p del cromosoma 21. Constituye un sistema de detoxificación natural que se expresa en varios tejidos humanos normales, asociados con funciones secretoras o de barrera, y actúa como una proteína transportadora de membrana responsable del eflujo celular de fármacos y tóxicos con peso molecular comprendido entre 300 y 2000 Da, entre los que se encuentran xenobióticos o fármacos tales como alcaloides de la vinca, verapamilo o corticosteroides, entre otros. La acción de glicoproteína-P parece ser doble, protegiendo a la célula del efecto de los fármacos e induciendo resistencia a la acción de éstos53-55. La sobreexpresión de glicoproteína-P se considera como uno de los mecanismos que motivan resistencia a quimioterápicos en enfermedades neoplásicas55 y se ha sugerido también su posible implicación en la resistencia a esteroides en enfermedades autoinmunes como el lupus eritematoso sistémico o la artritis reumatoide56-58. Recientemente, se ha demostrado que la IL-2 puede inducir un incremento en la expresión de ABCB1 y de glicoproteína-P, mediante la translocación del factor de transcripción específico, Y-box proteína-1, desde el citoplasma al núcleo de los linfocitos59. A través de este mecanismo, la IL-2 podría contribuir no sólo en la patogenia de la enfermedad por cambios mínimos, sino también en el desarrollo de resistencia a esteroides, sobre todo en enfermos que, por presentar múltiples brotes, se hallan expuestos de forma repetida y durante largos períodos de tiempo tanto a la acción de IL-2 a como a esteroides59.
En estudio reciente52, en el que se midió el nivel circulante de RR sIL-2 y se ha cuantificó la expresión de ABCB1 en linfocitos de enfermos con síndrome nefrótico secundario a NCM, se ha descrito que estos enfermos presentan unos niveles de RR sIL-2 y de ABCB1 mayores que los controles sanos, tanto durante el brote como tras la remisión. Durante la fase de brote, tanto los niveles de RR sIL-2 como los de ABCB1 fueron significativamente superiores en los enfermos con corticorresistencia y con múltiples brotes que en los enfermos en primer brote y en enfermos corticosensibles. Tras el tratamiento con esteroides, en los enfermos corticorresistentes los niveles de RR sIL-2 y los de ABCB1 no se modificaron significativamente. En los enfermos que respondieron, los niveles de RR sIL-2 y ABCB1 descendieron significativamente tras la remisión, pero en los enfermos con formas recidivantes permanecieron significativamente más elevados que en controles sanos. Estos datos coinciden con los publicados previamente por otros autores59-61 y sugieren que, en los enfermos con múltiples recidivas, que requieren tratamientos repetidos y prolongados con esteroides, la propia exposición al fármaco, asociada a la persistencia de activación de linfocitos T, podría estar implicada en el desarrollo de corticorresistencia o motivar la necesidad de dosis crecientes de esteroides para inducir el mismo efecto farmacológico. Los datos en los que se basa esta hipótesis son todavía escasos y requieren ser confirmados en estudios a mayor escala, pero tienen gran interés potencial en la medida en que podrían ser útiles para predecir la respuesta al tratamiento con esteroides, el riesgo de recidivas posteriores y/o aconsejar la indicación precoz de otras opciones de tratamiento en función de los niveles de RR sIL-2 y ABCB1 en el momento del diagnóstico o de la evolución de éstos durante el seguimiento.
Biomarcadores en la glomeruloesclerosis focal y segmentaria
La expresión «glomeruloesclerosis focal y segmentaria» se utiliza para definir una entidad que presenta un patrón de lesión definido en microscopía óptica, pero tiene múltiples posibles etiologías y patogenias62. La GFS se clasifica en primaria o secundaria en función de si se identifica o no una etiología responsable de ella63. Distinguir entre formas primarias y secundarias tiene un interés terapéutico y pronóstico trascendental, dado que sólo los enfermos con formas primarias que cursan con síndrome nefrótico y no son causadas por mutaciones en proteínas podocitarias son candidatos a tratamiento inmunosupresor o inmunomodulador64. En la actualidad, la diferenciación entre ambas se basa en el perfil clínico y el examen ultraestructural renal mediante microscopía electrónica. Aunque no es un criterio infalible, como norma, se considera que las formas primarias se caracterizan por presentar síndrome nefrótico y borramiento generalizado de los pies podocitarios en la microscopía electrónica. En las formas secundarias, puede observarse proteinuria de rango nefrótico, pero es inhabitual la presencia de síndrome nefrótico y el examen de microscopía electrónica permite demostrar que el borramiento de los pies podocitarios tiene una distribución focal y segmentaria en lugar de difusa65.
Los mecanismos patogénicos que causan lesión irreversible de los podocitos son poco conocidos tanto en las formas secundarias como en las primarias y, dentro de estas últimas, es altamente probable que no haya un mecanismo patogénico único común a todas ellas.
En los últimos años, la biología molecular ha permitido describir un número creciente de mutaciones que afectan a proteínas estructurales del podocito o proteínas integrantes del diafragma de filtración que pueden causar lesiones de GFS66,67. En la mayor parte de los casos, se trata de GFS que se inician en la infancia o en la adolescencia, habitualmente asociada a antecedentes familiares con diversos patrones de herencia y, en algunos casos, a síndromes extrarrenales que permiten tener la sospecha diagnóstica de que la lesión estructural renal se debe a la mutación de una proteína podocitaria. Sin embargo, aunque es un hecho infrecuente, hay evidencia de mutaciones esporádicas, sin historia familiar ni clínica extrarrenal asociada, en enfermos GFS que se inicia en edad adulta67. Estos datos indican que, aunque de manera muy inhabitual, algunas formas previamente consideradas primarias podrían ser debidas a mutaciones en proteínas podocitarias.
El hecho de que algunos enfermos con GFS primaria respondan a tratamiento con corticosteroides y/o inmunosupresores64,65 ha llevado a pensar que, en determinados casos, la patogenia puede estar relacionada con la activación de la respuesta inflamatoria y/o inmune, pero no se ha podido demostrar la existencia de fenómenos de autoinmunidad ni de disregulaciones de la respuesta inmunitaria con significado patogénico. La ausencia de depósitos inmunes en las biopsias, la evidencia de recidivas tras el trasplante renal que responden al tratamiento con plasmaféresis, inmunoadsorción o lipidoaféresis68-70 y la evidencia de transmisión del síndrome nefrótico de madre con GFS al recién nacido71 han aportado una base racional para generar la hipótesis sobre la existencia de un factor circulante o factor de permeabilidad (FP), capaz de lesionar el podocito27-29. La existencia de este factor (o factores) fue puramente especulativa hasta que se demostró que el plasma de algunos enfermos con GFS podía causar alteraciones en la permeabilidad a proteínas en glomérulos in vitro72. Desde entonces, el carácter patogénico del FP todavía no ha podido ser demostrado de forma indiscutible en los pacientes con formas primarias de GFS. En los casos en los que se ha identificado alguna molécula con características de FP, su presencia no se ha podido asociar de manera convincente ni con la respuesta al tratamiento ni con la recurrencia después del trasplante renal.
Niveles circulantes del receptor soluble de la uroquinasa
En fecha muy reciente se ha producido un avance potencialmente relevante al describirse que el nivel sérico del receptor soluble de uroquinasa (suPAR) está elevado en pacientes con GSF primaria, pero no en pacientes con otras enfermedades glomerulares73. El receptor de uroquinasa (uPAR) es un glicofosfatidilinositol capaz de transmitir señales intracelulares a través de su unión con integrinas de membrana74,75. Su función no es conocida, pero en modelos experimentales se ha demostrado que la inducción de señalización a través de uPAR en los podocitos causa fusión podocitaria y proteinuria a través de un mecanismo que depende de la activación de la alfaV beta3 integrina76. Por razones desconocidas, uPAR puede ser liberado de la membrana plasmática como forma soluble (suPAR)74,75. El suPAR tiene un peso molecular que oscila entre 20 y 50 kDa, similar al tamaño previsto para el hipotético FP descrito en algunos estudios77. En condiciones normales, sus concentraciones son bajas pero pueden estar elevadas en algunas neoplasias malignas, así como en individuos infectados por virus de la inmunodeficiencia humana78,79. Los datos disponibles indican que aproximadamente dos terceras partes de los enfermos con GFS primaria presentan niveles elevados de suPAR. En enfermos con GFS que reciben un trasplante renal, la presencia de un nivel elevado de suPAR previo al trasplante parece aumentar el riesgo de recidiva de la enfermedad en el riñón trasplantado y hay evidencia preliminar de que el tratamiento con plasmaféresis puede reducir significativamente los niveles e inducir la remisión73. A nivel experimental, se han podido inducir lesiones de GFS en ratones transgénicos que hiperexpresan suPAR73. Resultados experimentales del mismo grupo que ha descrito el aumento en los niveles circulantes de suPAR en enfermos con GFS primaria indican que suPAR podría ejercer su efecto mediante su unión a la β3 integrina podocitaria, una de las principales proteínas que sirve para anclar los podocitos a la membrana basal glomerular. La unión suPAR-β3 integrina causaría la activación del podocito y originaría cambios en su estructura y su función que alterarían la permeabilidad de la barrera de filtración glomerular. Aunque la identificación de la relación entre el incremento en los niveles de suPAR y la GFS es de gran trascendencia, ya que es la primera vez en la que es posible establecer una vinculación aparentemente consistente entre un factor circulante y la inducción de lesión podocitaria, todavía no se conoce qué células lo liberan a la circulación, qué factores regulan su síntesis ni cuáles son los motivos por los que los niveles de suPAR aumentan en un momento determinado y causan síndrome nefrótico. Por otra parte, debe destacarse que un alto porcentaje de enfermos con GFS no presentan niveles de suPAR circulante elevados. Se ha sugerido que en estos casos la lesión podocitaria podría producirse por señalización a través de uPAR local; sin embargo, también es posible que sea causada por mecanismos patogénicos no relacionados con esta vía. Por estas razones, para conocer cuál es el valor clínico de suPAR como posible biomarcador de GFS, es necesario realizar estudios con mayor número de enfermos y seguimiento prospectivo, para determinar qué niveles tienen valor diagnóstico y si la presencia de niveles elevados de suPAR guarda alguna relación con la presentación clínica, la respuesta al tratamiento o el pronóstico de la enfermedad. En caso de confirmarse, esta hipótesis abriría un nuevo camino para el estudio y tal vez para la orientación terapéutica de los enfermos con GFS, a la vez que supondría una nueva diana terapéutica ante la posibilidad de investigar intervenciones capaces de reducir los niveles de suPAR o de bloquear la unión suPAR-β3 integrina.
Biomarcadores, diagnóstico diferencial entre nefropatía por cambios mínimos y glomeruloesclerosis focal y segmentaria y predicción de la respuesta al tratamiento con esteroides
En el síndrome nefrótico idiopático causado por NCM o GFS la respuesta a esteroides ha sido identificada como la principal variable pronóstica a largo plazo, incluso independientemente del sustrato histológico, tanto en edad pediátrica como en adultos64,65. Si bien es cierto que la corticorresistencia se asocia con mayor frecuencia a patrón histológico de GFS, una parte de los enfermos con esta dolencia renal responde a esteroides o a otros inmunosupresores y la respuesta al tratamiento mejora significativamente su pronóstico64. Por otro lado, aunque la mayor parte de los enfermos con NCM responden a esteroides, hay enfermos con lesiones histológicas inequívocas de NCM que presentan corticorresistencia, bien al inicio, bien durante el curso evolutivo de la enfermedad. Esta ausencia de paralelismo estricto entre la presentación clínica o la lesión histológica y la respuesta al tratamiento ha motivado la realización de múltiples estudios orientados a buscar nuevos parámetros que permitan identificar a los enfermos, en función de su respuesta a los esteroides, desde el momento del diagnóstico. En la actualidad no hay ningún biomarcador que permita esta diferenciación, pero recientemente se han descrito algunos posibles candidatos. A nivel histológico, se ha descrito que el aumento en la expresión podocitaria de CD8019,20 y la reducción en la expresión de α-distroglicanos80,81 permitiría diferenciar la NCM de la GFS, pero no se ha descrito que un determinado perfil se asocie con respuesta a esteroides. Los niveles urinarios de CD8020 y de TGF-β82 han sido también propuestos como candidatos para diferenciar entre ambas entidades, pero tampoco se ha demostrado que se asocien con la respuesta al tratamiento. Además de la ya comentada relación entre expresión de ABCB1/glicoproteína-P y corticorresistencia52, se ha descrito una posible relación entre determinados polimorfismos en los genes que codifican la síntesis de IL-6, IL-4 y TNF-α y la respuesta a esteroides en niños con síndrome nefrótico idiopático83. Se han descrito, asimismo, perfiles proteómicos urinarios que serían distintos en función de la respuesta a esteroides, pero éstos todavía no se han evaluado en estudios clínicos84,85. En un estudio muy reciente86 se ha descrito la presencia de un fragmento de 13,8 KD de la α 1-B glicoproteína en la orina en el 36% de los enfermos corticorresistentes y en ninguno de los enfermos corticosensibles. La presencia de este fragmento de 1-B glicoproteína, sin embargo, se asocia a un menor filtrado glomerular y, por tanto, podría justificar la ausencia de respuesta por ser indicador de lesiones en estadios más avanzados. Estos datos, aunque de gran interés, deben ser confirmados en estudios clínicos más amplios.
Biomarcadores en la nefropatía membranosa
Nuevos autoanticuerpos en la nefropatía membranosa primaria. Valor diagnóstico, relación con la actividad clínica y con la respuesta al tratamiento
La NM es la primera causa de síndrome nefrótico idiopático en el adulto. Su base fisiopatológica consiste en la formación de depósitos inmunes en el espacio subepitelial, entre la lámina rara externa de la membrana basal glomerular y el podocito. La evidencia disponible indica que los depósitos se forman in situ en la cara basal de los procesos podocitarios, y posteriormente se desprenden y se fijan en el borde externo de la membrana basal glomerular87,88. La estructuración del modelo patogénico actualmente aceptado para la NM se ha fundamentado en su gran similitud con el modelo de nefritis experimental de Heymann89-92. Los hallazgos de la nefritis de Heymann nunca han podido ser reproducidos en el ser humano93,94, pero las evidencias aportadas por este modelo fueron la base para la realización de estudios con el objetivo de identificar el antígeno o los antígenos implicados en la patogenia de la NM humana.
Hasta hace poco, la única evidencia que relacionaba la presencia de un autoanticuerpo dirigido contra un antígeno podocitario con la aparición de síndrome nefrótico era la NM neonatal que aparece en hijos de madres con déficit de un antígeno podocitario denominado endopeptidasa neutra (NEP)95. Cuando éstas han sido inmunizadas contra dicho antígeno en embarazos previos, en nuevos embarazos los anticuerpos anti-NEP presentes en la sangre materna pasan a la circulación fetal a través de la barrera placentaria y, tras el anclaje con el antígeno presente en los podocitos del feto, forman inmunocomplejos y causan proteinuria.
Muy recientemente, utilizando homogeneizado de tejido renal sano y enfrentándolo a anticuerpos presentes en el suero de enfermos con NM primaria mediante técnicas de western-blot, seguidas de aislamiento e identificación del antígeno, se ha identificado el receptor tipo M de la fosfolipasa A2 (PLA2R) como primer antígeno podocitario diana de respuesta autoinmune en la NM primaria96.
El PLA2R presenta una organización estructural similar al receptor de manosa de los macrófagos y forma parte de un grupo de receptores de membrana que pertenece a la superfamilia de lectinas tipo C. El receptor humano fue originalmente clonado a partir de tejido renal, donde tiene un alto nivel de expresión a nivel de los podocitos97. Presenta un dominio extracelular grande, formado por la región N terminal rico en cisteína, un dominio tipo II fibronectina y una región de 8-10 AA de reconocimiento de carbohidratos distintos. Se considera que PLA2R transmite señales intracelulares tras su unión a una o varias de las fosfolipasas A2 solubles97-99. En modelos experimentales100, se ha demostrado que desempeña un papel importante en el shock endotóxico inducido por LPS, ya que los ratones que carecen del receptor tienen mayor resistencia a la acción de LPS. En el ser humano, su función no es conocida. La presencia de anticuerpos anti-PLA2R se considera específica de la NM primaria. Los datos de los estudios publicados hasta la fecha (tabla 2) indican que entre el 60 y el 70% de enfermos con NM primaria presentan niveles elevados de dichos anticuerpos96,101-103.
Tabla 2. Resumen de los resultados de los estudios clínicos y tipos de autoanticuerpos
descritos para el estudio de la nefropatía membranosa 
ACB: albúmina catiónica bovina; AR: aldosa reductasa; DB: dot blot; IF: inmunofluorescencia;
PLA2R: receptor tipo M de la fosfolipasa A2; SOD: superóxido-dismutasa 2; WB: western-blot.
Se ha descrito una clara correlación entre el título de anticuerpos (especialmente IgG4) y la actividad clínica de la enfermedad102,103 y se ha demostrado que el tratamiento con rituximab es capaz de reducir el título de anticuerpos, de forma paralela a la reducción en la excreción urinaria de proteínas104. Por otra parte, se ha identificado la presencia de depósitos de inmunocomplejos que contienen anticuerpos anti-PLA2R en la vertiente externa de la membrana basal glomerular, mediante estudios de inmunofluorescencia, incluso en enfermos en los que el nivel circulante de anticuerpos es negativo, lo que indicaría que un título de anticuerpos negativo (al menos con las técnicas disponibles en la actualidad) no permitiría excluir el diagnóstico de NM105. En enfermos trasplantados renales, la presencia de anticuerpos anti-PLA2R podría ser también de gran utilidad para diferenciar entre la recidiva de NM y la NM de novo postrasplante106. El papel de los anticuerpos anti-PLA2R en la patogénesis de la NM es desconocido. Se ha sugerido que la lesión podría producirse tras la formación de inmunocomplejos (anticuerpo/receptor) y la activación de complemento96. Se ha descrito susceptibilidad genética ligada a un alelo HLA-DQA1 localizado en 6p21107, de manera que los individuos homozigotos tendrían predisposición para la producción de anticuerpos no sólo frente al PLA2R, sino también frente a otros antígenos.
También se ha descrito una asociación entre determinados polimorfismos del PLA2R y riesgo de NM y, en el mismo estudio, se han descrito tanto haplotipos protectores como de riesgo para presentar la enfermedad pero no relacionados con el pronóstico108. El hecho de que los RR de PLA2 se hallen también presentes en otros lugares como pulmón o leucocitos indica que debe de haber otras variables locales que expliquen que el cuadro clínico se limite a la afección renal. Se ha identificado una forma soluble de PLA2R que se produce por splicing alternativo y que se supone que tendría funciones de regulación de la cantidad de fosfolipasa A2 libre circulante. Sin embargo, no se ha podido demostrar la existencia de niveles elevados de inmunocomplejos que contengan la forma soluble del receptor en enfermos con NM96, lo que se ha interpretado como una prueba a favor de la formación de inmunocomplejos in situ. La ausencia de anticuerpos frente a la forma soluble del receptor, cuyo peso molecular y estructura es distinta al receptor de membrana, coincide con la evidencia de que la inmunogenidad de la proteína requiere de la preservación de determinantes antigénicos conformacionales que sólo se hallan presentes en su estructura como proteína de membrana96. El hallazgo de anticuerpos de tipo IgG dirigidos contra el PLA2R en un elevado porcentaje de enfermos con NM primaria ha permitido desarrollar técnicas para medir sus niveles circulantes y tinciones específicas para detectar su presencia en las biopsias renales. Ambos procedimientos han supuesto un avance clínico de gran relevancia en el diagnóstico y seguimiento de los enfermos con NM, con potencialidad para facilitar la identificación de los enfermos con formas primarias o para proporcionar información sobre la actividad de la enfermedad y, de esta manera, orientar las decisiones terapéuticas. Aunque el valor diagnóstico, a la vista de los datos publicados, parece incuestionable, es necesario seguir ampliando el número de estudios para definir con claridad cuál es su valor pronóstico y su potencial utilidad como indicador precoz de recidiva.
La identificación en el hombre de un antígeno propio de la membrana del podocito contra el que actúan in situ anticuerpos tipo IgG indica que probablemente hay más antígenos locales implicados. Poco tiempo después de la identificación del PLA2R como diana de respuesta autoinmune, se han identificado dos nuevos autoanticuerpos frente a antígenos podocitarios, la aldosa-reductasa y la manganeso superóxido-dismutasa 2 (SOD2), que colocalizan con los depósitos de IgG y complemento y son selectivamente reconocidos por la IgG4 eluida del parénquima renal109. Estos anticuerpos parecen también específicos de la NM primaria, ya que no han podido ser demostrados en enfermos con formas secundarias ni en otras enfermedades renales. Su prevalencia no ha sido determinada en estudios clínicos y, hasta el momento, no se ha podido demostrar una vinculación etiopatogénica clara. Los estudios ultraestructurales localizan sus respectivos antígenos en el citoplasma de los podocitos y en los procesos podocitarios, y los datos disponibles indican que ambos antígenos son neoexpresados por parte de los podocitos, ya que, en el riñón sano, se localizan exclusivamente a nivel tubular. En el caso de la SOD2, hay datos in vitro que relacionan su expresión podocitaria con estrés oxidativo. El hecho de ser reconocidos por IgG4 los suma a la lista de nuevos antígenos identificados como posible diana de una respuesta autoinmune en la NM primaria. Sin embargo, no se conoce cuál es el factor que desencadena la neoexpresión antigénica ni cuál es la secuencia temporal de los hechos, de manera que, en la actualidad, no es posible determinar si la respuesta autoinmune observada es el desencadenante primario de la enfermedad o, de manera similar a lo que ocurre en otros procesos inflamatorios, es simplemente una consecuencia de una lesión del podocito causada por otros agentes de origen inmunológico o no, frente a la cual se desencadena una respuesta autoinmunitaria, de forma secundaria.
Los antígenos de origen extrarrenal son los responsables de la mayor parte de los casos de NM secundaria. Se considera que estos antígenos desencadenan la respuesta inmune tras su depósito a nivel extramembranoso, conducidos a través de la circulación y atravesando la membrana basal glomerular. Su origen puede ser múltiple, principalmente en relación con procesos autoinmunes sistémicos, infecciosos, neoplásicos o tras exposición a determinados fármacos y a antígenos alimentarios. Recientemente, se han descrito casos de NM en los que la lesión renal se produciría como consecuencia del depósito de inmunocomplejos formados por albúmina bovina e IgG antialbúmina bovina110.
Tinción para C4d en la biopsia renal
Aunque no se trata de un biomarcador circulante, en determinadas circunstancias el estudio de la vía de activación del complemento en las biopsias renales puede tener utilidad diagnóstica en el síndrome nefrótico causado por NM. La presencia de C4d en biopsias renales se considera una evidencia de la activación del complemento a través de la vía clásica o de la vía de las lectinas, pero no de la vía alternativa111. La utilidad de la tinción de C4d es ampliamente reconocida en el estudio de la patología del riñón trasplantado y, más recientemente, se ha extendido al estudio de las nefropatías primarias. La presencia de depósitos de C4d en biopsias de NM se conoce desde hace años112, pero no se ha evaluado su potencial aplicabilidad clínica hasta hace poco tiempo. En la NM que aparece en riñón nativo, el diagnóstico anatomopatológico no suele generar dudas debido al aspecto característico de la microscopía óptica, de la inmunofluorescencia y de la microscopía electrónica. Recientemente, se ha descrito la posibilidad de realizar tinción para C4d mediante técnicas de inmunohistoquimia en material parafinado113,114. En casos concretos en los que no se dispone de material suficiente para técnicas de inmunofluorescencia o de microscopía electrónica, la evidencia de tinción positiva para C4d en los capilares glomerulares puede ser útil para diferenciar la NM de la NCM y de la GFS cuando los datos de la microscopía óptica no son concluyentes. Por otra parte, en un estudio muy reciente, se ha descrito que en enfermos con NM que reciben un trasplante renal la positividad para C4d en capilares glomerulares en biopsias postrasplante puede ser un signo de recidiva de la nefropatía primaria que antecede a los cambios morfológicos característicos de la enfermedad y, en consecuencia, podría permitir el diagnóstico y tratamiento precoz115.
Proteinuria tubular como guía para la indicación de tratamiento inmunosupresor en la nefropatía membranosa
Tanto los criterios de indicación como el momento de inicio del tratamiento inmunosupresor en enfermos con NM primaria siguen siendo tema de debate, debido a la evidencia de que aproximadamente un 30-40% de los enfermos pueden presentar remisión espontánea, mientras que un porcentaje similar, si no recibe tratamiento, evoluciona hacia la insuficiencia renal116-119. Las variables clínicas de mal pronóstico han sido claramente identificadas, y la evolución espontánea, tanto en un sentido como en otro, suele ser evidente en los primeros 2 o 3 años de seguimiento. En las guías clínicas120, habitualmente se aconseja un período de espera bajo tratamiento conservador, incluyendo bloqueantes de angiotensina II, antes de indicar tratamiento inmunosupresor. Por consenso, se suele recomendar que la duración mínima de este período sea de 6 meses, pero la duración debe individualizarse en cada caso en función del número de factores de mal pronóstico presentes.
Sin embargo, la evidencia en la que se basan estas recomendaciones no es suficientemente sólida como para que no haya autores que la cuestionen y argumenten que, incluso considerando los períodos de observación recomendados en las guías, hay riesgo de exponer a muchos pacientes innecesariamente a la toxicidad de los inmunosupresores119. Estos grupos plantean limitar el tratamiento inmunosupresor a los pacientes con mayor riesgo de sufrir insuficiencia renal progresiva116,121. Aunque se ha demostrado que esta estrategia es posible con altas tasas de supervivencia renal121,122, la dificultad está en consensuar cuál es el mejor parámetro para identificar a los pacientes de alto riesgo.
Los defensores de la restricción terapéutica, inicialmente, consideraron que la evidencia de deterioro de la función renal era el marcador más específico para indicar tratamiento116,123,124. Sin embargo, el aplazamiento del inicio del tratamiento hasta la evidencia de deterioro de la función renal116,121 puede favorecer la progresión de las lesiones renales hacia la fibrosis, limitar la eficacia del tratamiento, causar respuestas incompletas o insuficiencia renal residual. En este contexto, se ha justificado buscar marcadores subrogados que permitan la predicción del pronóstico en fase temprana de la enfermedad y orienten la decisión terapéutica, antes de que haya deterioro de la función renal. La mayor parte de los datos disponibles al respecto se centran en analizar la capacidad de los niveles urinarios de proteinuria tubular como marcador subrogado precoz del deterioro de función renal125-129. Branten et al.126 analizaron la validez y la precisión de la excreción urinaria de beta-2 microglobulina (β2m) e IgG como predictores de la aparición de insuficiencia renal, definida por un ascenso en la creatinina superior al 50% o una creatinina sérica superior a 1,5 mg/dl, en una cohorte de enfermos con NM y función renal basal normal. Los enfermos recibieron tratamiento inmunosupresor sólo cuando hubo evidencia de empeoramiento de la función renal. El 44% de los enfermos presentó deterioro de la función renal, cifra que coincide con la publicada en estudios previos sobre evolución espontánea de enfermos no tratados. En todos los casos, el empeoramiento se produjo durante los primeros 36 meses de seguimiento. Un nivel de excreción urinaria de β2m ≥ 0,5µg/min e IgG ≥ 250 mg/24 h se asoció a riesgo de progresión con igual sensibilidad y con mayor especificidad y valor predictivo positivo que la proteinuria, por lo que los autores proponen que la medición de dichos parámetros podría ser de ayuda para decidir iniciar o no tratamiento inmunosupresor. Sin embargo, el valor de estos datos en la práctica clínica es muy difícil de precisar, ya que no se realizó un análisis multivariado para identificar los predictores independientes de progresión, ni se describe la variabilidad de los niveles urinarios de β2m e IgG cuando se miden en un mismo enfermo a lo largo del tiempo. Por otra parte, de acuerdo con los datos del estudio, basar la decisión terapéutica en los niveles de β2m o IgGimplicaría no tratar al 10% de los enfermos en los que finalmente se deteriora la función renal.
Se ha sugerido que los niveles urinarios de α1-microglobulina y de otras proteínas de bajo peso molecular podrían tener un valor predictivo similar127, pero ninguna de ellas ha sido validada en estudios cínicos independientes. La medición de β2m plantea problemas en la práctica clínica diaria porque sólo puede realizarse si el pH urinario es superior a 6, ya que en orina ácida se degrada. Por ello, se han analizado otras proteínas tubulares de comportamiento más estable. Se ha descrito una buena relación entre la excreción urinaria de β-N-acetil glucosaminidasa (NAG), cuyo nivel no depende del pH urinario, y el pronóstico en enfermos con NM128, y en un estudio reciente129, en el que se compara el valor predictivo de β2m con el de NAG, se concluye que, aunque ambas pueden ser útiles en la predicción del pronóstico, β2m es más precisa. En dicho estudio, en el análisis multivariado, la excreción urinaria de β2m fue el mejor predictor independiente de deterioro de función renal.
En conjunto, aunque carecen de validación externa, estos datos sugieren que los niveles urinarios de β2m y NAG podrían ser útiles para el seguimiento de los enfermos durante la fase de observación previa a la decisión terapéutica. La presencia de unos niveles de β2m o NAG elevados se asociaría a mal pronóstico y podría ser una variable a considerar para tomar la decisión de iniciar tratamiento inmunosupresor. Sin embargo, la ausencia de datos sobre la variabilidad de los niveles de β2m y NAG, cuando se miden en un mismo individuo a lo largo del tiempo, junto a la evidencia de que aproximadamente el 15% de los enfermos con niveles de β2m bajos presentan deterioro de la función renal, y casi un 20% de los enfermos con niveles de β2m elevados pueden entrar en remisión espontánea, dificultan definir la utilidad clínica real de estos parámetros. Por otra parte, considerando el filtrado glomerular basal de los enfermos (71 ± 23 ml/min/1,73 m2), es obvio que en dicho estudio se incluyó a enfermos que presentaban función renal basal reducida. Este hecho dificulta la valoración de la capacidad de β2m y NAG para predecir la evolución de la función renal antes de que ésta se deteriore -puesto que en algunos casos ya lo ha hecho- y también dificulta la extrapolación de los datos del estudio a los enfermos con función renal normal. Además, la presencia de enfermos con función renal basal reducida puede tener gran importancia a la hora de interpretar los predictores independientes de la evolución de la función renal descritos, ya que tanto β2m como NAG, en la medida en que reflejan extensión de las lesiones tubulointersticiales130, pueden ser indicadores de la presencia de lesiones renales más avanzadas. Por ello, es probable que los enfermos con niveles más elevados de β2m y NAG tuvieran también menor filtrado glomerular inicial y, en consecuencia, menor probabilidad de remisión espontánea o de respuesta al tratamiento.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Referencias Bibliográficas
1. Rivera F, Alcázar R, Egido J, Peces R, Pérez-García R, Praga M, et al. Síndrome nefrótico. In: Rodríguez Pérez JC, Orte Martínez LM (eds.). Normas de Actuación Clínica en Nefrología. Madrid: Harcourt Brace de España; 1998. p 19-28. [ Links ]
2. Alcázar R, Egido J. Síndrome nefrótico. Fisiopatología y tratamiento general. In: Hernando L (ed.). Nefrología Clínica. 2.a ed. Madrid: Panamericana; 2004. p. 277-289. [ Links ]
3. Koomans HA. Patophysiology of oedema in idiopathic nephritic syndrome. Nephrol Dial Transplant 2003;18 Suppl 6:vi30-2. [ Links ]
4. International Study of Kidney Disease in Children. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. J Pediatr 1981;98:561-4. [ Links ]
5. Adu A. The nephrotic syndrome: does renal biopsy affect management? Nephrol Dial Transplant 1996;11:12-4. [ Links ]
6. Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet 1974;ii:556-60. [ Links ]
7. Grimbert P, Audard V, Remy P, Lang P, Sahali D. Recent approaches to the pathogenesis of minimal-change nephritic syndrome. Nephrol Dial Transplant 2003;18:245-8. [ Links ]
8. Mathieson PW. Immune disregulation in minimal change nephropathy. Nephrol Dial Transplant 2003;18 Suppl 6:vi26-vi29. [ Links ]
9. Brenchley PE. Vascular permeability factors in steroid-sensitive nephrotic syndrome and focal segmental glomeruloesclerosis. Nephrol Dial Transplant 2003;18 Suppl 6:vi21-vi25. [ Links ]
10. Boner G, Cox AJ, Kelly DJ, Tobar A, Bernheim J, Langham RG, et al. Does vascular endothelial growth factor (VEGF) play a role in the pathogenesis of minimal change disease? Nephrol Dial Transplant 2003;18:2293-99. [ Links ]
11. Musante L, Candiano G, Zennaro C, Bruschi M, Carraro M, Artero M, et al. Humoral permeability factors in the nephrotic syndrome: a compendium and prospectus. J Nephrol 2001;14 Suppl 4:S48-50. [ Links ]
12. Yap HK, Cheung W, Murugasu B, Sim SK, Seah CC, Jordan SC. Th1 and Th2 cytokine mRNA profiles in childhood nephrotic syndrome: Evidence for increased IL-13 mRNA expression in relapse. J Am Soc Nephrol 1999;10:529-37. [ Links ]
13. Koyama A, Fujisaki M, Kobayashi M, Igarashi M, Narita M. A glomerular permeability factor produced by human T cell hybridomas. Kidney Int 1991;40:453-60. [ Links ]
14. Hoxha E, Stahl RA, Harendza S. Rituximab in adult patients with immunosuppressive-dependent minimal change disease. Clin Nephrol 2011;76:151-8. [ Links ]
15. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest 2004;113:1390-7. [ Links ]
16. Reiser J, Mundel P. Danger signaling by glomerular podocytes defines a novel function of inducible B7-1 in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol 2004;15:2246-8. [ Links ]
17. Chambers CA, Allison JP. Costimulatory regulation of T cell function. Curr Opin Cell Biol 1999;11:203-10. [ Links ]
18. Shimada M, Ishimoto T, Lee PY, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, et al. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-{kappa}B-dependent pathway. Nephrol Dial Transplant 2012;27:81-9. [ Links ]
19. Lai KW, Wei CL, Tan LK, Tan PH, Chiang GS, Lee CG, et al. Overexpression of interleukin-13 induces minimal-change-like nephropathy in rats. J Am Soc Nephrol 2007;18:1476-85. [ Links ]
20. Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, et al. Urinary CD80 excretion increases in idiopathic minimal-change disease. J Am Soc Nephrol 2009;20:260-6. [ Links ]
21. Garin EH, Mu W, Arthur JM, Rivard CJ, Araya CE, Shimada M, et al. Urinary CD80 is elevated in minimal change disease but not in focal segmental glomerulosclerosis. Kidney Int 2010;78:296-302. [ Links ]
22. Kapp U, Yeh WC, Patterson B, Elia AJ, Kagi D, Ho A, et al. Interleukin 13 is secreted by andstimulates the growth of Hodgkin and Reed-Sternberg cells. J Exp Med 1999;189:1939-46. [ Links ]
23. Skinnider BF, Elia AJ, Gascoyne RD, Trumper LH, von Bonin F, Kapp U, et al. Interleukin 13 and interleukin 13 receptor are frequently expressed by Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2001;98:2877-8. [ Links ]
24. Skinnider BF, Kapp U, Mak TW. Interleukin 13: A growth factor in Hodgkin lymphoma. Int Arch Allergy Immunol 2001;126:267-76. [ Links ]
25. Cheung W, Wei CL, Seah CC, Jordan SC, Yap HK. Atopy, serum IgE, and interleukin-13 in steroid-responsive nephrotic syndrome. Pediatr Nephrol 2004;19:627-32. [ Links ]
26. Chen SP, Cheung W, Heng CK, Jordan SC, Yap HK. Childhood nephrotic syndrome in relapse is associated with down-regulation of monocyte CD14 expression and lipopolysaccharide-induced tumour necrosis factor-alpha production. Clin Exp Immunol 2003;134:111-9. [ Links ]
27. Wei CL, Cheung W, Heng CK, Arty N, Chong SS, Lee BW, et al. Interleukin-13 genetic polymorphisms in Singapore Chinese children correlate with long-term outcome of minimal-change disease. Nephrol Dial Transplant 2005;20:728-34. [ Links ]
28. van den Berg JG, Aten J, Anwar Chand M, Claessen N, Dijkink L, Wijdenes J, et al. Interleukin-4 and interleukin-13 act on glomerular visceral epithelial cells. J Am Soc Nephrol 2000;11:413-22. [ Links ]
29. Lai KW, Wei CL, Tan LK, Tan PH, Chiang GS, Lee CG, et al. Overexpression of interleukin-13 induces minimal-change-like nephropathy in rats. J Am Soc Nephrol 2007;18:1476-85. [ Links ]
30. Kimata H, Fujimoto M, Furusho K. Involvement of interleukin (IL)-13, but not IL-4, in spontaneous IgE and IgG4 production in nephrotic syndrome. Eur J Immunol 1995;25:1497-1501. [ Links ]
31. Kuzelova K, Mrhalova M, Hrkal Z. Kinetics of heme interaction with heme-binding proteins: the effect of heme aggregation state. Biochim Biophys Acta 1997;1336:497-501. [ Links ]
32. Vincent SH, Grady RW, Shaklai N, Snider JM, Muller-Eberhard U. The influence of heme-binding proteins in hemecatalyzed oxidations. Arch Biochem Biophys 1988;265:539-50. [ Links ]
33. Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995;41:1819-28. [ Links ]
34. Immenschuh S, Nagae Y, Satoh H, Baumann H, Muller-Eberhard U. The rat and human hemopexin genes contain an identical interleukin-6 response element that is not a target of CAAT enhancer-binding protein isoforms. J Biol Chem 1994;269:12654-61. [ Links ]
35. Immenschuh S, Song DX, Satoh H, Muller-Eberhard U. The type II hemopexin interleukin-6 response element predominates the transcriptional regulation of the hemopexin acute phase responsiveness. Biochem Biophys Res Commun 1995;207:202-8. [ Links ]
36. Kapojos JJ, Poelstra K, Borghuis T, Banas B, Bakker WW. Regulation of plasma hemopexin activity by stimulated endothelial or mesangial cells. Nephron Physiol 2004;96:P1-10. [ Links ]
37. Kapojos JJ, van den Berg A, van Goor H, Te Loo MWM, Poelstra K, Borghuis T, et al. Production of hemopexin by TNFa stimulated human mesangium cells. Kidney Int 2003;63:1681-6. [ Links ]
38. Cheung PK, Stulp B, Immenschuh S, Borghuis T, Baller JF, Bakker WW. Is 100KF an isoform of hemopexin? Immunochemical characterization of the vasoactive plasma factor 100 KF. J Am Soc Nephrol 1999;10:1700-8. [ Links ]
39. Cheung PK, Klok PA, Baller JF, Bakker WW. Induction of experimental proteinuria in vivo following infusion of humanplasma hemopexin. Kidney Int 2000;57:1512-20. [ Links ]
40. Bakker WW, Van Dael C, Pierik LJ, van Wijk JA, Nauta J, Borghuis T, et al. Altered activity of plasma hemopexin in patients with minimal change disease in relapse. Pediatr Nephrol 2005;20:1410-5. [ Links ]
41. Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol 1993;11:245-68. [ Links ]
42. Rubin LA, Kurman CC, Fritz ME, Biddison WE, Boutin B, Yarchoan R, et al. Soluble interleukin-2 receptors are released by activated lymphocytes in vitro. J Immunol 1985;135:3172-7. [ Links ]
43. Rubin LA, Nelson DL. The soluble interleukin-2 receptor: biology, function and clinical application. Ann Intern Med 1990;113:619-27. [ Links ]
44. Ohno I, Gomi H, Matsuda H,Nakano H, Matsumoto H, Kodama K, et al. Soluble IL-2 receptor in patients with primary nephritic syndrome. Nihon Jinzo Gakkai Shi 1991;33:483-9. [ Links ]
45. Mandreoli M, Beltrandi E, Casadei-Maldini M, Mancini R, Zucchelli A, Zucchelli P. Lymphocyte release of soluble IL-2 receptors in patients with minimal change nephropathy. Clin Nephrol 1992;37:177-82. [ Links ]
46. Bock GH, Ongkingco JR, Patterson LT, Ruley J, Schroepfer LR, Nelson DL. Serum and urine soluble interleukin-2 receptor in idiopathic nephrotic syndrome. Pediatr Nephrol 1993;7:523-8. [ Links ]
47. Hulton SA, Shah V, Byrne MR, Morgan G, Barratt TM, Dillon MJ. Lymphocyte subpopulations, interleukin-2 and interleukin-2 receptor expression in childhood nephrotic syndrome. Pediatr Nephrol 1994;8:135-9. [ Links ]
48. Chen HS, Wu MS, Yen TS, Chen WY. Soluble interleukin-2 receptor in patients with glomerular diseases. Postgrad Med J. 1995;71:617-22. [ Links ]
49. Ayli MD, Duman N, Duranay M, Ates K, Ayli M, Karatan O, et al. Serum levels of soluble interleukin-2 receptor in patients with primary nephrotic syndrome. Nephron 1998;80:349-50. [ Links ]
50. Pogan A, Sancewicz-Pach K, Miezynski W. TNF-alpha and soluble interleukin-2 receptor and glomerular sclerosis in primary nephrotic syndrome in children. Wiad Lek 2005;58 Suppl 1:39-44. [ Links ]
51. Kemper MJ, Meyer-Jark T, Lilova M, Müller-Wiefel DE. Combined T- and B-cell activation in childhood steroid-sensitive nephrotic syndrome. Clin Nephrol 2003;60:242-7. [ Links ]
52. Youssef DM, Elbehidy RM, Abdelhalim HS, Amr GE. Soluble interleukine-2 receptor and MDR1 gene expression levels as inflammatory biomarkers forprediction of steroid response in children with nephrotic syndrome. Iran J Kidney Dis 2011;5:154-61. [ Links ]
53. Bradley G, Juranka PF, Ling V. Mechanism of multidrug resistance. Biochim Biophys Acta 1988;948:87-128. [ Links ]
54. Meijer OC, de Lange EC, Breimer DD, de Boer AG, Workel JO, de Kloet ER. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology 1998;139:1789-93. [ Links ]
55. Masters JR. Biochemical basis of resistance to chemotherapy. Radiother Oncol 1990;19:297-305. [ Links ]
56. Webster JI, Carlstedt-Duke J. Involvement of multidrug resistance proteins (MDR) in the modulation of glucocorticoid response. J Steroid Biochem Mol Biol 2002;82:277-88. [ Links ]
57. Tsujimura S, Saito K, Nakayamada S, Nakano K, Tanaka Y. Clinical relevance of the expression of P-glycoprotein on peripheral blood lymphocytes to steroid resistance in patients with systemic lupus erythematosus. Arthritis Rheum 2005;52:1676-83. [ Links ]
58. Maillefert JF, Maynadie M, Tebib JG, Aho S, Walker P, Chatard C, et al. Expression of the multidrug resistance glycoprotein 170 in the peripheral blood lymphocytes of rheumatoid arthritis patients. The percentage of lymphocytes expressing glycoprotein 170 is increased in patients treated with prednisolone. Br J Rheumatol 1996;35:430-5. [ Links ]
59. Wasilewska AM, Zoch-Zwierz WM, Pietruczuk M. Expression of P-glycoprotein in lymphocytes of children with nephrotic syndrome treated with glucocorticoids. Eur J Pediatr 2006;165:839-44. [ Links ]
60. Wasilewska A, Zoch-Zwierz W, Pietruczuk M. Expression of multidrug resistance P-glycoprotein on lymphocytes from nephrotic children treated with cyclosporine A and ACE-inhibitor. Eur J Pediatr 2007;166:447-52. [ Links ]
61. Funaki S, Takahashi S, Wada N, Murakami H, Harada K. Multiple drug-resistant gene 1 in children with steroid sensitive nephrotic syndrome. Pediatr Int 2008;50:159-61. [ Links ]
62. Quereda C, Ballarín J. Síndrome nefrótico por glomerulosclerosis focal segmentaria del adulto. Nefrologia 2007;27 (Suppl 2):56-69. [ Links ]
63. Appel AS, D'Agati VD. Primary and secondary (non-genetic) causes of focal and segmental glomerulosclerosis. In: Floege J, Johnson RJ, Feehally J (eds.). Comprehensive clinical nephrology (4th ed.). St. Louis: Elsevier Saunders; 2010. p. 228-40. [ Links ]
64. Meyrier A. Management of idiopathic nephrotic syndrome in adults: minimal change disease and focal segmental glomerulosclerosis. In: Molony DA, Craig JC, eds. Evidence-based nephrology (4th ed.). Oxford: Wiley-Blackwell; 2009. p. 149-57. [ Links ]
65. Praga M. Tratamiento de la glomeruloesclerosis segmentaria y focal. Nefrologia 2005;25:612-21. [ Links ]
66. Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 2006;354:1387-401. [ Links ]
67. Santín S, Bullich G, Tazón-Vega B, García-Maset R, Giménez I, Silva I, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2011;6:1139-48. [ Links ]
68. Hickson LJ, Gera M, Amer H, Iqbal CW, Moore TB, Milliner DS, et al. Kidney transplantation for primary focal segmental glomerulosclerosis: outcomes and response to therapy for recurrence. Transplantation 2009;87:1232-39. [ Links ]
69. Artero ML, Sharma R, Savin VJ, Vincenti F. Plasmapheresis reduces proteinurianand serum capacity to injure glomeruli in patients with recurrent focal glomerulosclerosis. Am J Kidney Dis 1994;23:574-81. [ Links ]
70. Haas M, Godfrin Y, Oberbauer R, Yilmaz N, Borchhardt K, Regele H, et al. Plasma immunadsorption treatment in patients with primary focal and segmental glomerulosclerosis. Nephrol Dial Transplant 1998;13:2013-6. [ Links ]
71. Kemper MJ, Wolf G, Müller-Wiefel DE. Transmission of glomerular permeability factor from a mother to her child. N Engl J Med 2001;344:386-7. [ Links ]
72. McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2010;5:2115-21. [ Links ]
73. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 2011;17:952-60. [ Links ]
74. Blasi F, Carmeliet P. uPAR: a versatile signaling orchestrator. Nat Rev Mol Cell Biol 2002;3:932-43. [ Links ]
75. Smith HW, Marshall CJ. Regulation of cell signaling by uPAR. Nat Rev Mol Cell Biol 2010;11:23-36. [ Links ]
76. Wei C, Möller CC, Altintas MM,Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med 2008;14:55-63. [ Links ]
77. Sharma M, Sharma R, McCarthy ET, Savin VJ. "The FSGS factor": enrichment and in vivo effect of activity from focal segmental glomerulosclerosis plasma. J Am Soc Nephrol 1999;10:552-61. [ Links ]
78. Sier CF, Stephens R, Bizik J, Mariani A, Bassan M, Pedersen N, et al. The level of urokinase-type plasminogen activator receptor is increased in serum of ovarian cancer patients. Cancer Res 1998;58:1843-9. [ Links ]
79. Sidenius N, Sier CF, Ullum H, Pedersen BK, Lepri AC, Blasi F, et al. Serum level of soluble urokinase-type plasminogen activator receptor is a strong and independent predictor of survival in human immunodeficiency virus infection. Blood 2000;96:4091-5. [ Links ]
80. Regele HM, Fillipovic E, Langer B, Poczewki H, Kraxberger I, Bittner RE, et al. Glomerular expression of dystroglycans is reduced in minimal change nephrosis but not in focal segmental glomerulosclerosis. J Am Soc Nephrol 2000;11:403-12. [ Links ]
81. Giannico G, Yang H, Neilson EG, Fogo AB. Dystroglycan in the diagnosis of FSGS. Clin J Am Soc Nephrol 2009;4:1747-53. [ Links ]
82. Woroniecki RP, Shatat IF, Supe K, Du Z, Kaskel FJ. Urinary cytokines and steroid responsiveness in idiopathic nephrotic syndrome of childhood. Am J Nephrol 2008;28:83-90. [ Links ]
83. Tripathi G, Jafar T, Mandal K,Mahdi AA, Awasthi S, Sharma RK, et al. Does cytokine gene polymorphism affect steroid responses in idiopathic nephrotic syndrome? Indian J Med Sci 2008;62:383-91. [ Links ]
84. Woroniecki RP, Orlova TN, Mendelev N, Shatat IF, Hailpern SM, Kaskel FJ, et al. Urinary proteome of steroid-sensitive and steroid-resistant idiopathic nephrotic syndrome of childhood. Am J Nephrol 2006;26:258-67. [ Links ]
85. Traum AZ. Urine proteomic profiling to identify biomarkers of steroidresistance in pediatric nephrotic syndrome. Expert Rev Proteomics 2008;5:715-9. [ Links ]
86. Piyaphanee N, Ma Q, Kremen O, Czech K, Greis K, Mitsnefes M, et al. Discovery and initial validation of alpha 1-B glycoprotein fragmentation as a differential urinary biomarker in pediatric steroid-resistant nephrotic syndrome. Proteomics Clin Appl 2011;5:334-42. [ Links ]
87. Kerjaschki D. Pathomechanisms and molecular basis of membranous glomerulopathy. Lancet 2004;364:1194-6. [ Links ]
88. Ronco P, Debiec H. Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol 2005;16:1205-13. [ Links ]
89. Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JLP. Production of nephrotic syndrome in rats by Freund´s adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med 1959;100:660-4. [ Links ]
90. Leung CC, Cheewatrakoolpong B, O'Mara T, Black M. Passive Heymann nephritis induced by rabbit antiserum to membrane antigens isolated from rat visceral yolk-sac microvilli. Am J Anat 1987;179:169-74. [ Links ]
91. Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci U S A 1982;79:5557-61. [ Links ]
92. Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol 1995;6:35-47. [ Links ]
93. Collins AB, Andres GA, McCluskey RT. Lack of evidence for a role of renal tubular antigen in human membranous glomerulonephritis. Nephron 1981;27:297-301. [ Links ]
94. Whitworth JA, Leibowitz S, Kennedy MC, Cameron JS, Evans DJ, Glassock RJ, et al. Absence of glomerular renal tubular epithelial antigen in membranous glomerulonephritis. Clin Nephrol 1976;5:159-62. [ Links ]
95. Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med 2002;346:2053-60. [ Links ]
96. Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009;361:11-21. [ Links ]
97. Ancian P, Lambeau G, Mattéi MG, Lazdunski M. The human 180-kDa receptor for secretory phospholipases A2. Molecular cloning, identification of a secreted soluble form, expression, and chromosomal localization. J Biol Chem 1995;270:8963-70. [ Links ]
98. Lambeau G, Ancian P, Nicolas JP, Cupillard L, Zvaritch E, Lazdunski M. A family of receptors for secretory phospholipases A2. C R Seances Soc Biol Fil 1996;190:425-35. [ Links ]
99. Zvaritch E, Lambeau G, Lazdunski M. Endocytic properties of the M-type 180-kDa receptor for secretory phospholipases A2. J Biol Chem 1996;271:250-7. [ Links ]
100. Hanasaki K, Yokota Y, Ishizaki J, Itoh T, Arita H. Resistance to endotoxic shock in phospholipase A2 receptor-deficient mice. J Biol Chem 1997;272:32792-7. [ Links ]
101. Qin W, Beck LH Jr, Zeng C, Chen Z, Li S, Zuo K, et al. Anti-phospholipase A2 receptor antibody in membranous nephropathy. J Am Soc Nephrol 2011;22:1137-43. [ Links ]
102. Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ. Anti-phospholipase Areceptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 2011;6:1286-91. [ Links ]
103. Hoxha E, Harendza S, Zahner G, Panzer U, Steinmetz O, Fechner K, et al. An immunofluorescence test for phospholipase-A2-receptor antibodies and its clinical usefulness in patients with membranous glomerulonephritis. Nephrol Dial Transplant 2011;26:2526-32. [ Links ]
104. Beck LH Jr, Fervenza FC, Beck DM, Bonegio RG, Malik FA, Erickson SB, et al. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol 2011;22:1543-50. [ Links ]
105. Debiec H, Ronco P. PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N Engl J Med 2011;364:689-90. [ Links ]
106. Debiec H, Martin L, Jouanneau C, Dautin G, Mesnard L, Rondeau E, et al. Autoantibodies specific for the phospholipase A2 receptor in recurrent and De Novo membranous nephropathy. Am J Transplant 2011;11:2144-52. [ Links ]
107. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 2011;364:616-26. [ Links ]
108. Liu YH, Chen CH, Chen SY, Lin YJ, Liao WL, Tsai CH, et al. Association of phospholipase A2 receptor 1 polymorphisms with idiopathic membranous nephropathy in Chinese patients in Taiwan. J Biomed Sci 2010;17:81. [ Links ]
109. Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol 2010;21:507-19. [ Links ]
110. Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschênes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med 2011;364:2101-10. [ Links ]
111. Brown KM, Sacks SH, Sheerin NS. Mechanisms of disease: the complement system in renal injury--new ways of looking at an old foe. Nat Clin Pract Nephrol 2007;3:277-86. [ Links ]
112. Kusunoki Y, Itami N, Tochimaru H, Takekoshi Y, Nagasawa S, Yoshiki T. Glomerular deposition of C4 cleavage fragment (C4d) and C4-binding protein in idiopathic membranous glomerulonephritis. Nephron 1989;51:17-9. [ Links ]
113. Lopez M, Espinosa M, Ortega R, Gomez JM, Perez MJ, Toiledo K, et al. C4d como herramienta en el diagnóstico de la nefropatía membranosa. Nefrologia 2009;29Suppl 2:6. [ Links ]
114. Val-Bernal JF, Garijo MF, Val D, Rodrigo E, Arias M. C4d immunohistochemical staining is a sensitive method to confirm immunoreactant deposition in formalin-fixed paraffin-embedded tissue in membranous glomerulonephritis. Histol Histopathol 2011;26:1391-7. [ Links ]
115. Rodriguez EF, Cosio FG, Nasr SH, Sethi S, Fidler ME, Stegall MD, et al. The pathology and clinical features of early recurrent membranous glomerulonephritis. Am J Transplant 2012;12:1029-38. [ Links ]
116. du Buf-Vereijken PW, Branten AJW, Wetzels JFM. Idiopathic membranous nephropathy: outline and rationale of a treatment strategy. Am J Kidney Dis 2005;46:1012-29. [ Links ]
117. Cattran DC. Management of membranous nephropathy: when and what for treatment. J Am Soc Nephrol 2005;16:1188-94. [ Links ]
118. Schiepatti A, Mosconi L, Perna A, Mecca G, Bertani T, Garattini S, et al. Prognosis of untreated patients with idiopathic membranous nephropathy. N Engl J Med 1993;329:85-9. [ Links ]
119. Perna A, Schiepatti A, Zamora J, Giuliano GA, Braun N, Remuzzi G. Immunosuppressive treatment for idiopathic membranous nephropathy: a systematic review. Am J Kidney Dis 2004;44:385-401. [ Links ]
120. Fulladosa X, Praga M, Segarra A, Martínez Ara J. Glomerulonefritis membranosa. Nefrologia 2007;27 Suppl 2:70-86. [ Links ]
121. Branten AJ, Reichert LJ, Koene RA, Wetzels JF. Oral cyclophosphamide versus chlorambucil in the treatment of patients with membranous nephropathy and renal insufficiency. QJM 1998;91:359-66. [ Links ]
122. du Buf-Vereijken PW, Feith GW, Hollander DA, Gerlag PG, Wirtz JJ, Noordzij TC, et al. Restrictive use of immunosuppressive treatment in patients with idiopathic membranous nephropathy: high renal survival in a large patient cohort. QJM 2004;97:353-60. [ Links ]
123. Reichert LJM, Koene RAP, Wetzels JFM. Prognostic factors in idiopathic membranous nephropathy. Am J Kidney Dis 1998;31:1-11. [ Links ]
124. Honkanen E, Törnroth T, Grönhagen-Riska C, Sankila R. Long-term survival in idiopathic membranous glomerulonephritis: can the course be clinically predicted? Clin Nephrol 1994;41:127-34. [ Links ]
125. Reichert LJ, Koene RA, Wetzels JF. Urinary excretion of beta 2-microglobulin predicts renal outcome in patients with idiopathic membranous nephropathy. J Am Soc Nephrol 1995;6:1666-9. [ Links ]
126. Branten AJW, du Buf-Vereijken PW, Klasen IS, Bosch FH, Feith GW, Hollander DA, etal. Urinary excretion of b2-microglobulin and IgG predict prognosis in idiopathic membranous nephropathy: a validation study. J Am Soc Nephrol 2005;16:169-74. [ Links ]
127. Bazzi C, Petrini C, Rizza V, Arrigo G, Beltrame A, Pisano L, et al. Urinary excretion of IgG and a1- microglobulin predicts clinical course better than extent of proteinuria in membranous nephropathy. Am J Kidney Dis 2001;38:240-8. [ Links ]
128. Bazzi C, Petrini C, Rizza V, Arrigo G, Napodano P, Paparella M, et al. Urinary N-acetyl-beta-glucosaminidase excretion is a marker of tubular cell dysfunction and a predictor of outcome in primary glomerulonephritis. Nephrol Dial Transplant 2002;17:1890-6. [ Links ]
129. Bazzi C, Petrini C, Rizza V, Arrigo G, Beltrame A, D'Amico G. Characterization of proteinuria in primary glomerulonephritides. SDS-PAGE patterns: clinical significance and prognostic value of low molecular weight ("tubular") proteins. Am J Kidney Dis 1997;29:27-35. [ Links ]
130. Portman RJ, Kissane JM, Robson AM, Peterson LJ, Richardson A. Use of beta2-microglobulin to diagnose tubulointerstitial renal lesions in children. Kidney Int 1986;30:91-8. [ Links ]
Enviado a Revisar: 20 Abr. 2012 ![]() Dirección para correspondencia:
Dirección para correspondencia:
Alfonso Segarra-Medrano,
Servicio de Nefrología,
Hospital Universitari Vall d'Hebron, Barcelona
E-mail: alsegarr@gmail.com
Aceptado el: 11 Jun. 2012

