










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNefrología (Madrid)
On-line version ISSN 1989-2284Print version ISSN 0211-6995
Nefrología (Madr.) vol.33 n.3 Cantabria 2013
https://dx.doi.org/10.3265/Nefrologia.pre2012.Nov.11735
Plasmaféresis en el tratamiento del síndrome hemolítico-urémico típico
Plasmapheresis in the treatment of typical haemolytic-uraemic syndrome
Dirección para correspondencia
Sr. Director:
El síndrome hemolítico urémico (SHU) es un trastorno de la microvasculatura, clínicamente definido por anemia hemolítica microangiopática (negativa en el test de Coombs) y trombocitopenia, que afecta preferentemente a los riñones manifestándose con hematuria, oligoanuria y fracaso renal1.
Caso clínico
Mujer de 14 años sin antecedentes personales de interés. Tras el regreso de un viaje a Estambul, comienza con episodio de deposiciones diarreicas de características mucosanguinolentas (14-15 deposiciones/día) que se acompañan de vómitos, dolor abdominal, debilidad generalizada y febrícula. Tras la persistencia de esta clínica durante cinco días, comienza con hematuria y oligoanuria acudiendo a nuestro hospital, donde analíticamente se detecta deterioro del filtrado glomerular (creatinina: 1,62 mg/dl), plaquetopenia (84.000 mm3) y anemia hemolítica acompañada de hiperbilirrubilemia a expensas de bilirrubina directa (BD) (hemoglobina: 10,6 g/dl, BD: 2,3 mg/dl), estudio de coagulación sin alteraciones. Ante la sospecha de SHU, se realiza nuevo control analítico con frotis sanguíneo, apreciándose empeoramiento severo tanto clínica como analíticamente: creatinina: 2,6 mg/dl, plaquetas: 39.000 mm3, hemoglobina: 7 g/dl (precisó transfusión de 2 concentrados de hematíes). Destaca la presencia de esquistocitos en el frotis sanguíneo, siendo la realización de coprocultivo negativo para toxina Shiga.
Se realizó estudio inmunológico con inmunoglobulinas, complemento, anticuerpos anticitoplasma de neutrófilos, anticuerpos antinucleares, anticuerpo antimembrana basal glomerular, que resultó negativo, descartándose por tanto enfermedad sistémica.
Ante estos datos, se diagnostica de SHU y se inician medidas de soporte con sueroterapia y sesión de plasmaféresis a través de catéter transitorio femoral derecho, y se añade al tratamiento prednisona (1 mg/kg/peso).
Durante los siete primeros días, la evolución fue tórpida sin respuesta a sesiones de plasmaféresis diaria y con empeoramiento de la clínica comenzando con descenso brusco del recuento plaquetario (19.000/mm3) y fracaso renal agudo con anuria (función renal: creatinina: 6 mg/dl). Llegó a precisar 3 sesiones de hemodiálisis.
En la undécima sesión de plasmaféresis diaria, comienza con aumento progresivo del número de plaquetas (86.000/mm3), así como mejoría de la función renal (creatinina: 1,5 mg/dl) y aumento del ritmo de diuresis (3000 cc/24 h) suspendiéndose las sesiones de hemodiálisis.
Dada la gran mejoría clínica y analítica, se decide continuar con sesiones de plasmaféresis a días alternos con progresiva mejoría tanto de función renal (hasta normalizarse [creatinina: 1,1 mg/dl]) y de la anemia hemolítica (hemoglobina: 9,6 g/dl) como del aumento del recuento plaquetario (plaquetas: 115.000/mm3);diuresis 24 h/2500 cc. La evolución de la paciente se resume en la figura 1.
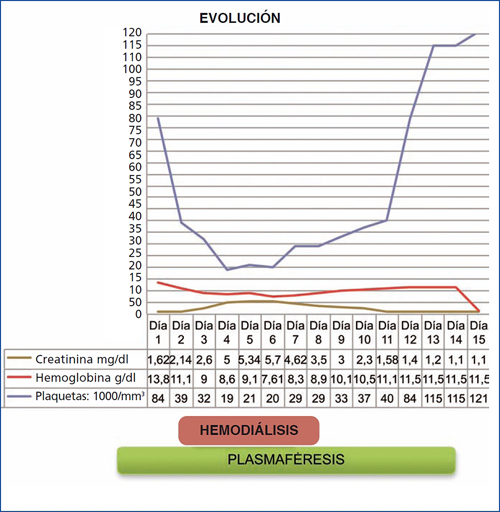
Figura 1. Gráfica de evolución
En la actualidad, la paciente se encuentra clínicamente asintomática, normotensa y con parámetros analíticos (función renal, hemoglobina y plaquetas) dentro de la normalidad.
Discusión
El SHU es una constelación de signos y síntomas caracterizado por la tríada anemia hemolítica microangiopática, trombocitopenia y fracaso renal agudo.
Este trastorno puede dividirse en dos formas en función de la asociación o no con bacterias que producen la toxina Shiga-like, así como de la presentación clínica:
- SHU típico o asociado a toxina Shiga (Stx): es la forma más común del SHU (90 % de los casos). Está causada por la infección transmitida por los alimentos con toxina Shiga producida por Escherichia coli. Por lo general, transcurre después de un episodio prodrómico de diarrea, con frecuencia sanguinolenta. Suele ser autolimitada y normalmente tiene un curso benigno2.
- SHU atípico o no asociado a toxina Shiga: representa un 10 % de los casos de SHU. La enfermedad puede aparecer de forma esporádica (< 20 % de los casos) o de origen familiar. Es un grupo heterogéneo de trastornos que se distingue clínicamente por la ausencia de diarrea en el 92 % de los casos y peor pronóstico. La mayoría de los pacientes presenta recurrencias y más de un 50 % desarrollan enfermedad renal crónica estadio 53.
El diagnóstico para establecer la asociación entre SHU e infección por E. coli productor de toxina Shiga (STEC) se basa en tres criterios: aislamiento y caracterización del patógeno; detección de Stx libre en materia fecal y detección de anticuerpos anti-Stx en suero4.
Una de las limitaciones para la aplicación de todos los enfoques terapéuticos nuevos y específicos es que la ventana de tiempo entre el diagnóstico de infección por STEC y la aplicabilidad de estos es muy estrecha. El diagnóstico de la infección por STEC debería realizarse dentro de las 48 h posteriores a la iniciación de la diarrea5.
En nuestro caso, no fue posible la confirmación de diagnóstico dado que la paciente acudió seis días tras el inicio de la clínica; no obstante, dada la clínica sugerente de infección por E. coli (dolor abdominal acompañado de deposiciones diarreicas sanguinolentas) junto con los hallazgos de trombopenia, hemólisis microangiopática (elevación de LDH y disminución de la hemoglobina) y aparición de fracaso renal agudo, así como la evolución favorable de la paciente, confirmamos que estábamos ante un caso de SHU típico.
Lo primordial ante una sospecha de SHU es el inicio rápido de tratamiento, ya que por lo general sigue un curso progresivo produciendo un fracaso renal irreversible, deterioro neurológico progresivo, isquemia cardíaca e incluso la muerte
Concluimos insistiendo en que la mejor forma de disminuir los casos de SHU es la prevención: controles más estrictos en todos los puntos de la cadena alimentaria que aseguren el cumplimiento de las leyes y normativas de control bromatológico en todos los niveles, junto con políticas educativas dirigidas a toda la población5.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
M. Inmaculada Poveda-García, M. Carmen Prados-Soler, M. Dolores Del Pino-y-Pino,
Remedios Garófano-López, Mercedes Alfaro-Tejeda, Francisco J. Guerrero-Camacho,
Beatriz García-Maldonado, David Sánchez-Martos y Felisa Sánchez-Martínez
Unidad de Gestión Clínica de Nefrología. Hospital Torrecárdenas. Almería
Referencias Bibliográficas
1. Gruppo RA. Managing Atypcal Hemolytic-Uremic Syndroe: What does the future hold? Disponible en http://www.medscape.org/viewarticle/751918Mescape education 2011. [ Links ]
2. Ake JA, Jelacic S, Ciol MA, Watkins SL, Murray KF, Christie DL, et al. Relative nephroprotection during Escherichia coli 0157:H7 infections: association with intravenous volume expansion. Pediatrics 2005;115:e673-680. [ Links ]
3. Rodríguez de Córdoba S, Montes T. Síndrome hemolítico urémico atípico. Nefrologia Sup Ext 2011;2(1):58-65. [ Links ]
4. Fagundo J, Delgado Y, Síndrome Hemolítico Urémico Rev Cubana Hematol Inmuno Hemoter 2003;19(2-3). [ Links ]
5. Fernández-Brando RJ, Bentancor LV, Mejías MP. Actualización en el tratamiento del síndrome urémico hemolítico endémico patogéncesis y tratamiento de la complicación sistémica más grave de las infecciones por Escherichia Coli productor de toxina Shiga. Medicina 2011;71:383-9. [ Links ]
6. Gianantonio C, Vitacco M, Mendilaharzu F, Rutty A, Mendilaharzu J. The Hemolytic-Uremic Syndrome. J Pedriatric 1964;64:478-91. [ Links ]
7. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009;361:1676-87. [ Links ]
8 Chiurchiu C, Remuzzi G. Síndrome hemolítico urémico. Nefrologia 2003;23 Suppl 3:13-20. [ Links ]
9. Fakhouri F, Vernant JP, Veyradier A, Wolf M, Kaplanski G, Binaut R, et al. Efficienciy of curative and prophylactic treatment with rituximab in ADAMTS 13 - deficient thrombotic thrombocytopenic purpura: a study of 11 cases. Blood 2005;106:1932. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
M. Inmaculada Poveda-García
Unidad de Gestión Clínica de Nefrología
Hospital Torrecárdenas, Sagrada Familia portal 10, 2o A
04006, Almería
inmapoved@hotmail.com

