










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.33 no.4 Cantabria 2013
https://dx.doi.org/10.3265/Nefrologia.pre2013.Apr.12006
CASOS CLÍNICOS
Síndrome uña-rótula. Un caso con una mutación de novo en el gen LMX1B no descrita previamente
Nail-patella syndrome. A case with a de novo mutation in the LMX1B gene not previously described
Naira Álvarez-Martín1, María J. Gamundi2, Imma Hernan2, Miguel Carballo2, M. Isabel Luis-Yanes1, Víctor García-Nieto1
1Sección de Nefrología Pediátrica. Servicio de Pediatría. Hospital Nuestra Señora de Candelaria. Santa Cruz de Tenerife
2Unidad de Genética Molecular. Hospital de Terrassa. Terrassa, Barcelona
Dirección para correspondencia
El síndrome uña-rótula es una enfermedad hereditaria infrecuente producida por mutaciones que originan una pérdida de función del factor de transcripción LMX1B. La prevalencia se estima en 1/50000 recién nacidos. Se transmite con un patrón de herencia autosómica dominante con una penetrancia completa. El gen pleiotrópico LMX1B, miembro de la familia de genes «homeo» implicados en el desarrollo, interviene en la configuración normal del eje dorsoventral y de la membrana basal glomerular durante el desarrollo embrionario.
La enfermedad fue descrita por primera vez por Little en 18971, aunque la descripción clásica de la enfermedad fue realizada por Fong en 19462. La asociación con enfermedad renal fue establecida por Hawkins y Smith en 19503. El gen responsable fue descrito por Dreyer et al. en 19984.
La gravedad del fenotipo clínico es muy variable. Básicamente, se trata de una enfermedad con afectación de las uñas, el sistema esquelético, los riñones y los ojos. Las manifestaciones clínicas que definen la enfermedad se agrupan en la tétrada clínica consistente en displasia ungueal (98%), hipoplasia o aplasia de las rótulas (74%), limitación funcional de los codos (70%) y la presencia de cuernos ilíacos (70%); estos últimos, que son patognomónicos, pueden observarse mediante ecografía desde el tercer trimestre de gestación. Alrededor del 40% de los pacientes presentan afectación renal consistente en hematuria y proteinuria. Un 5-10% desarrollan proteinuria en rango nefrótico en la niñez o adolescencia y progresan a insuficiencia renal terminal durante períodos variables de tiempo. Con el microscopio electrónico se visualiza borramiento de los pedicelos de los podocitos, una membrana basal glomerular ensanchada con zonas de rarefacción, y depósitos dispersos de fibrillas de colágeno, que también están incrementados en la matriz mesangial. El factor de transcripción LMX1B se expresa en la vida posnatal en el podocito, lo que sugiere un papel regulador de diversos genes en esta célula, como NPHS2 y CD2AP5.
Hemos tenido la oportunidad de estudiar a una niña de 8 años y 3 meses de edad que fue remitida por el hallazgo de proteinuria. Al nacimiento, destacó el bajo peso al nacer, junto con la presencia de rasgos dismórficos, como pies zambos, artrogriposis leve en codos, macrocefalia e hipoplasia ungueal. La niña adquirió la marcha a los 21 meses, debido a una displasia de caderas. Fue sometida a varias intervenciones quirúrgicas para corregir sus defectos esqueléticos. En la exploración clínica actual se apreció una baja estatura, macrocefalia con frente muy prominente, sinofridia y cejas muy pobladas, filtrum nasal ancho con labio superior fino, hendiduras antimongoloides, narinas antevertidas, hipertelorismo, ausencia de ambas rótulas (figura 1) e hipoplasia ungueal de los dedos de las manos con uñas distróficas (figura 2). No se objetivaron edemas. Es la segunda hija de unos padres no consanguíneos, sin antecedentes de enfermedad renal en ninguna de las ramas familiares. El padre presenta alteraciones leves en las uñas. En los exámenes complementarios se comprobó proteinuria en rango nefrótico (7,3 g/l; cociente proteínas/creatinina [Cr]: 6,73 mg/mg), hipoproteinemia (5,3 g/l), reducción de los niveles de IgG (512 mg/dl) e hipercolesterolemia (312 mg/dl). Filtrado glomerular renal (FGR) normal (134 ml/min/1,73 m2). El estudio del gen LMX1B mostró la mutación en heterocigosis c.728G>C (p.Trp243Ser). Esta mutación no ha sido descrita previamente en la literatura. Dado que las mutaciones del gen LMX1B solo han sido relacionadas con el síndrome uña-rótula, asumimos que es la responsable del cuadro clínico de la paciente. En el estudio genético realizado a los familiares de primer grado no se observó la mutación, por lo que debe de tratarse de una mutación de novo. Se instauró tratamiento con enalapril oral, en un intento de reducir la proteinuria, y simvastatina. En el último control, la proteinuria ha disminuido a 5,35 g/l (cociente proteínas/Cr: 2,84 mg/mg), el FGR se mantiene normal y la paciente sigue libre de edemas.
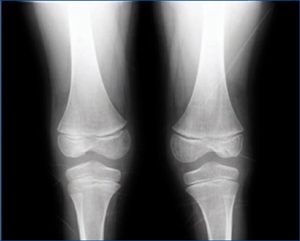
Figura 1. Ausencia de rótulas

Figura 2. Hipoplasia ungueal y uñas distróficas
En esta enfermedad, se han descrito más de 130 mutaciones diferentes consistentes, en su mayoría, en cambios de un solo nucleótido. Están distribuidas predominantemente entre los exones 2 y 6. Se han descrito una serie de mutaciones más frecuentes que, en su conjunto, representan el 30% del total. Un 12% de las mutaciones son de novo7. No se ha establecido una correlación fenotipo-genotipo, por lo que, aunque se puede realizar el diagnóstico prenatal, existe una marcada variabilidad inter e intrafamiliar7. La confirmación de una mutación en el gen LMX1B evita la realización de una biopsia renal para confirmar el diagnóstico. No existe un tratamiento específico. Las lesiones de la membrana basal glomerular no recidivan tras la realización de un trasplante renal.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Referencias bibliográficas
1. Little EM. Congenital absence or delayed development of the patella. Lancet 1897;2:781-4. [ Links ]
2. Fong EE. Iliac horns (symmetrical bilateral central posterior iliac processes). Radiology 1946;47:517. [ Links ]
3. Hawkins CF, Smith OE. Renal dysplasia in a family with multiple hereditary abnormalities including iliac horns. Lancet 1950;1:803-8. [ Links ]
4. Dreyer SD, Zhou G, Baldini A, Winterpacht A, Zabel B, Cole W, et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 1998;19:47-50. [ Links ]
5. Lemley KV. Kidney disease in nail-patella syndrome. Pediatr Nephrol 2009;24:2345-54. [ Links ]
6. McIntosh I, Dunston JA, Liu L, Hoover-Fong JE, Sweeney E. Nail Patella syndrome revisited: 50 years after linkage. Ann Hum Genet 2005;69:349-63. [ Links ]
7. Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 2003;40:153-62. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Naira Álvarez-Martín,
Sección de Nefrología Pediátrica.
Servicio de Pediatría,
Hospital Nuestra Señora de Candelaria,
Ctra. General del Rosario s/n,
38010, Santa Cruz de Tenerife
naivila@hotmail.com
vgarcianieto@gmail.com
Enviado a Revisar: 10 Mar. 2013
Aceptado el: 11 Abr. 2013

