










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNefrología (Madrid)
On-line version ISSN 1989-2284Print version ISSN 0211-6995
Nefrología (Madr.) vol.33 n.5 Cantabria 2013
https://dx.doi.org/10.3265/Nefrologia.pre2013.Jun.11190
Síndrome de Bardet-Biedl, modelo de ciliopatía e importancia del compromiso renal
Bardet-Biedl syndrome, the ciliopathy model and the importance of renal involvement
Dirección para correspondencia
Sr. Director:
Bardet en 1920 y Biedl en 1922 describieron un síndrome congénito caracterizado por obesidad, polidactilia, retinitis pigmentosa, retraso mental e hipoplasia gonadal. Otras anormalidades asociadas frecuentemente son hipertensión y diabetes mellitus1,2. Los pacientes adultos consultan al nefrólogo por hipertensión, pruebas de imagen renal anómalas o insuficiencia renal, siendo esta la mayor causa de morbimortalidad1.
Epidemiología y diagnóstico
La incidencia varía geográficamente entre 1/160000 en Suiza y 1/13500 entre beduinos de Kuwait, probablemente asociado a endogamia2. El diagnóstico es básicamente clínico. Beales et al. en 20011 sugieren que la presencia de cuatro criterios mayores o tres mayores asociados a dos menores pueda ser diagnóstico (tabla 1), pero este se puede retrasar debido a emergencia lenta y variabilidad clínica en la expresión de la enfermedad. Los diagnósticos diferenciales del cuadro clínico de un niño obeso, con retraso psicomotor, sin polidactilia, incluyen una amplia serie de posibilidades, hasta que se asocie una alteración visual3.
Tabla 1. Criterios diagnósticos del síndrome de Bardet-Biedl (Beales et al.). 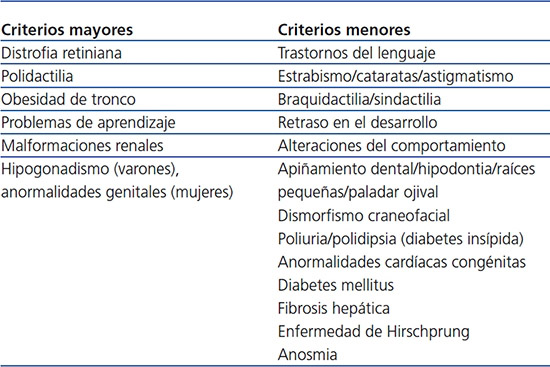
Genética del Síndrome de Bardet-Biedl
Se han estudiado al menos 16 genes relacionados con el síndrome de Bardet-Biedl (SBB), entre ellos BBS1, BBS2, ARL6/BBS3 y SDCCAG8/BBS16. En el modo de herencia autosómico recesivo, los padres son heterocigotos obligados y asintomáticos, los hermanos tendrán una probabilidad del 25% de ser sanos, 50% serán heterocigotos y 25% afectados por la enfermedad. La herencia trialélica implica la presencia de al menos tres mutaciones para que se manifieste el fenotipo. Actualmente se desconoce hasta qué punto el multialelismo contribuye al fenotipo1,4.
Malformaciones renales y función renal alterada
El compromiso renal se observa hasta en el 40% de los casos. La displasia renal se caracteriza por alteraciones en el parénquima y nefronoptisis. En la ecografía y la resonancia magnética nuclear se encuentran lobulaciones. El cuadro clínico incluye anemia, poliuria y polidipsia en la niñez. Menos frecuente es la enfermedad glomerular con glomeruloesclerosis focal y segmentaria y despegamiento de la membrana basal glomerular, aunque rara vez se realiza una biopsia renal5.
Tratamiento
No existe tratamiento para la pérdida de visión, pero se puede prever la educación especial y el entrenamiento futuro para la ceguera. La obesidad se debe manejar con dieta y ejercicio. La dislipemia y la hipertensión se tratan igual que en la población general y, según el grado de la insuficiencia renal, se realizará seguimiento especializado. Se debe ofrecer consejo genético1,6.
Caso clínico
Mujer de 50 años, sin alergias, intervenida quirúrgicamente por polidactilia a los dos años de edad. Retinitis pigmentaria diagnosticada hace más de 25 años. Enfermedad pulmonar obstructiva crónica tipo bronquitis crónica, fumadora activa de 20 paquetes/año. Niega ceguera en familiares de primer y segundo grado. Exploración física: peso: 85,9 kg, talla: 1,65 m, índice de masa corporal: 31,55 kg/m2. Analítica de sangre sin anemia, sin dislipemia ni aumento de la creatinina. Estudio genético en la Fundación Jiménez Díaz de Madrid en mayo de 2010: portadora de una mutación en el gen BBS1 que es responsable de su enfermedad. Ecografía abdominal (figura 1): hígado graso, páncreas no valorable, bazo homogéneo. Riñón derecho con suaves lobulaciones y quiste sinusal de 3,8 cm, riñón izquierdo con prominencia cortical redondeada, probable lobulación, sin poder descartar tumoración renal, por lo que aconsejan resonancia magnética nuclear.

Figura 1. Ecografía abdominal que muestra lobulaciones en ambos riñones.
Síndrome de Bardet-Biedl como ciliopatía
Los cilios están presentes en casi todas las células de los vertebrados. Intervienen en el desarrollo embrionario, en la polaridad y la homeostasis, en funciones sensoriales (oído, vista, olfato) y transportadoras, y en la división celular. Su ensamblaje y mantenimiento depende del transporte intraflagelar que lleva partículas del cuerpo basal a lo largo de la estructura microtubular hasta la punta. En 2003 Ansley et al.7 propusieron que la patogénesis molecular del SBB se debía al compromiso ciliar. Muchas proteínas codificadas por genes alterados en el SBB se localizan en el centrosoma y el cuerpo basal. Las células glomerulares y tubulares producen un único cilio de estructura 9 + 0 que se proyecta a la luz del túbulo y funciona como mecano o quimiorreceptor. Otras ciliopatías incluyen el síndrome de Kartagener, la enfermedad poliquística AD y la nefronoptisis (figura 2).

Figura 2. El cilio.
Conclusiones
El SBB es una enfermedad genética muy rara. Aunque clínicamente está bien definida y caracterizada por la asociación de polidactilia, retinitis pigmentaria, retraso psicomotor y obesidad, genéticamente es heterogénea, incluyendo genes que codifican proteínas para el funcionamiento y la estructura de los cilios, por lo que este síndrome se agrupa dentro de las ciliopatías, que como entidades patogénicas han despertado un renovado interés científico. Revisando la literatura médica, encontramos que en España se han descrito 4 casos en total, que corresponden a 2 familias con 2 hermanos afectos cada una9,10.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
M. Isabel Acosta-Ochoa1, Karina Ampuero-Anachuri1, Ruth Tavarez-Paniagua2,
M. Eugenia Plagaro-Cordero1, Antonio Molina-Miguel1
1Servicio de Nefrología. Hospital Universitario Río Hortega. Valladolid
2Medicina familiar y Comunitaria. Área Oeste, Valladolid.
Referencias Bibliográficas
1. Forsythe E, Beales PL. Bardet-Biedl syndrome. European Journal of Human Genetics (2013) 21, 8-13. [ Links ]
2. O'Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis 1996;27(6):776-83. [ Links ]
3. Waters AM, Beales PL. Bardet-Biedl Syndrome, GeneReviews (Internet). Pagon RA, Bird TD, Dolan CR, et al., editors. Seattle (WA): University of Washington, Seattle; 1993. Initial Posting: July 14, 2003; Last Revision: November 18, 2010. [ Links ]
4. Beales PL. Lifting the lid on Pandora's Box: the Bardet-Biedl syndrome. Curr Opin Genet Dev 2005;15:315-23. [ Links ]
5. Putoux A, Attie-Bitach T, Martinovic J, Gubler MC. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol 2012;27(1):7-15. [ Links ]
6. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 2011;26:1039-56. [ Links ]
7. Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003;425(6958):628-33. [ Links ]
8. Dollfus H, Verloes A, Bonneau D, Cossée M, Perrin-Schmitt F, Brandt C, et al. Le point sur le syndrome de Bardet-Biedl. J Fr Ophtalmol 2005;28(1):106-12. [ Links ]
9. Carrasco MA, Ayuso C, Inglada L, Grant C, Herrera P. Síndrome de Bardet-Biedl: algunas peculiaridades de su presentación. Rev Clin Esp 1988;182:26-9. [ Links ]
10. Merino Angulo J, Barrio Arredondo MT, Cartón Trigo F. Síndrome de Bardet-Biedl: alteraciones renales subclínicas en 2 hermanos. An Med Interna 1992;9(10):493-4. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
M. Isabel Acosta-Ochoa,
Servicio de Nefrología,
Hospital Universitario Río Hortega,
macostao@saludcastillayleon.es

