










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.34 no.5 Cantabria 2014
https://dx.doi.org/10.3265/Nefrologia.pre2014.Jun.12379
REVIEWS
Serum calcium and bone: effect of PTH, phosphate, vitamin D and uremia
Calcio sérico y huesos: efectos de la hormona paratiroidea, del fosfato, de la vitamina D y de la uremia
Barton S. Levine1, Mariano Rodríguez2 and Arnold J. Felsenfeld1
1Departments of Medicine. VA Greater Los Angeles Healthcare System and the David Geffen School of Medicine at UCLA. Los Angeles, CA (USA)
2Department of Nephrology. Red in Ren. IMIBIC. Hospital Universitario Reina Sofía. Córdoba (Spain)
M.R. has received grant support from the Instituto Carlos III (FIS 07/0287, FIS 07/0315, 010/1311), Consejería de Salud (JA 0127/2008), a UE Grant from Framework Programme 7 Syskid (FP7-241544), and Consejería de Innovación, Ciencia y Empresa (CTS-5205 and CTS-170) of Junta de Andalucía.
ABSTRACT
Hyperparathyroidism develops in chronic kidney disease (CKD). A decreased calcemic response to parathyroid hormone (PTH) contributes to the development of hyperparathyroidism and is presumed due to reduced calcium efflux from bone. Contributing factors to the decreased calcemic response to PTH in CKD include: 1) hyperphosphatemia; 2) decreased serum calcitriol; 3) downregulation of the PTH1 receptor; 4) large, truncated amino-terminal PTH fragments acting at the carboxy-PTH receptor; and 5) uremic toxins. Also, prolonged high dose calcitriol administration may decrease the exchangeable pool of bone calcium independent of PTH. The goal of the review is to provide a better understanding of how the above cited factors affect calcium efflux from bone in CKD. In conclusion, much remains to be learned about the role of bone in the regulation of serum calcium.
Key words: Bone, Calcium, Parathyroid hormone, Phosphate, Uremia, Vitamin D.
RESUMEN
El hiperparatiroidismo se desarrolla en la enfermedad renal crónica (ERC). La disminución de la respuesta calcémica a la hormona paratiroidea (PTH) contribuye al desarrollo de hiperparatiroidismo y es probable que se deba a una reducción de la emisión de calcio de los huesos. Entre los factores que contribuyen a la disminución de la respuesta calcémica a la PTH en la ERC se encuentran: 1) la hiperfosfatemia; 2) la disminución del calcitriol sérico; 3) la desensibilización del receptor PTHR1; 4) la presencia de fragmentos de gran tamaño de los extremos aminoterminales de la hormona paratiroidea que actúan en el receptor carboxi-PTH y 5) las toxinas urémicas. Asimismo, la administración prolongada de una dosis elevada de calcitriol podría disminuir la reserva intercambiable de calcio independiente de la hormona paratiroidea. El objetivo de esta revisión es facilitar la comprensión de cómo afectan los factores mencionados anteriormente a la emisión de calcio procedente del hueso en la ERC. Como conclusión, aún queda mucho por aprender acerca del papel de los huesos en la regulación del calcio sérico.
Palabras clave: Hueso, Calcio, Hormona paratiroidea, Fosfato, Uremia, Vitamina D.
The decreased calcemic response to parathyroid hormone (PTH) in chronic kidney disease (CKD)1,2 challenges us to evaluate how calcium is mobilized from bone. Hyperparathyroidism develops in CKD to prevent hypocalcemia. Factors contributing to the need for increased PTH include: 1) hyperphosphatemia or even inappropriately normal serum phosphorus for an elevated PTH; 2) decreased serum calcitriol; 3) downregulation of the PTH1 receptor; 4) accumulation of large, truncated amino-terminal PTH fragments such as 7-84 PTH; and 5) uremic toxins. A review of how these factors affect the calcemic response to PTH in CKD is instructive.
Parathyroid hormone
The relationship between PTH and serum calcium is a sigmoidal curve with basal PTH approximately 25% of the maximal PTH response to hypocalcemia.3 During hypocalcemia, increased PTH secretion restores serum calcium to normal. Besides its bone effect, PTH increases renal calcium reabsorption and calcitriol production enhancing gut absorption of calcium. However, the instantaneous effect and that of greatest magnitude is attributed to calcium efflux from bone.
Not often discussed is the role of a calcium gradient with greater mobilization of calcium from bone in hypocalcemia than in hypercalcemia. When increasing PTH doses were infused in parathyroidectomized rats, the serum calcium response was curvilinear with a steeper response between 5 and 10mg/dL than at higher values (Figure 1 A) showing that the calcium gradient in hypercalcemia reduced the calcemic action of PTH.4 The effect of infused PTH on serum phosphorus was also curvilinear with a steeper decrease at lower PTH doses (Figure 1 B). These results show an important role for a calcium gradient in mobilizing calcium from bone with a possible complementary effect for phosphorus lowering.

Interestingly, even in the absence of PTH, calcium efflux from bone does occur in response to reductions in serum calcium. In parathyroidectomized, hypocalcemic rats, the rate of recovery from ethylene glycol tetraacetic acid (EGTA) induced lowering of serum calcium to baseline hypocalcemic values was similar to that in parathyroid-intact rats EGTA-induced hypocalcemia to normocalcemia.5 In dogs, recovery from hypocalcemia was shown to be more rapid in young than in adult dogs, probably due to a greater exchangeable pool of bone calcium in young animals.6 Results from animal studies suggest that the increase in calcium is bone derived.5,7,8
What is the short-term role of PTH and does PTH play a role in maintaining the constancy of serum calcium throughout the day? Clinical studies have shown that daily variations in serum calcium values are greater in hypoparathyroid patients than in normal humans and are affected more by variations in dietary calcium and phosphorus.9 A possible explanation is that during fasting, the normal human is better able to extract calcium from bone and during feeding to better deposit calcium into bone to maintain a constant serum calcium concentration.10,11
In the early 1990s, D'Amour and associates showed that during PTH stimulation from hypocalcemia, the ratio of intact PTH to carboxy (C)-PTH fragments increased and during PTH suppression from hypercalcemia, the ratio of intact PTH to C-PTH fragments decreased.12 Subsequently, the same authors showed that besides 1-84 PTH, the assay for intact PTH also detected non 1-84 PTH large truncated amino-terminal fragments of which 7-84 PTH was the prototype.13 In normal humans, non 1-84 PTH large truncated amino-terminal fragments represent about 20% of measured intact PTH, but in renal failure, these same fragments account for approximately 50%.12,13 Interest increased when the 7-84 PTH fragment was shown to inhibit the calcemic action of 1-84 and 1-34 PTH.8,14 The action of 7-84 PTH is most likely mediated by its binding to the C-PTH receptor present on osteocytes and other cells of osteoblast lineage including lining cells on the bone surface.15,16 Moreover, the osteocyte has important signaling functions to the bone surface.17 Finally, 7-84 PTH may internalize the PTH1 receptor in selected target cells in kidney and bone18 and also reduce calcitriol production.19 Thus, in advanced renal failure, greatly increased 7-84 PTH could decrease the calcemic response to PTH in absence of hypercalcemia.2
An intriguing observation in azotemic rats is that parathyroidectomy completely restored the calcemic response to PTH even though hyperphosphatemia was present before the 1-34 PTH infusion (Figure 2 A).20 Before the PTH infusion, rats were maintained on a high calcium diet for more than two weeks after parathyroidectomy. It is possible that the total absence of 7-84 PTH or other PTH fragments resulted in the greater response to PTH. Other possibilities not mutually exclusive include that the total absence of PTH resulted in an upregulation of the PTH1 receptor, hypocalcemia lessened the gradient driving mineralization increasing the exchangeable bone pool of calcium, and the high calcium diet resulted in expansion of the exchangeable bone pool of calcium.

The size of the exchangeable pool of bone calcium differs among the different forms of renal osteodystrophy in CKD. In studies in hemodialysis patients performed in the 1980s and 1990s, we used low and high calcium dialysates during hemodialysis to induce hypo- and hypercalcemia to evaluate PTH stimulation and suppression. The capacity to defend against the development of hypocalcemia and hypercalcemia was much greater in hemodialysis patients with high (osteitis fibrosa) than with low bone turnover (adynamic bone or osteomalacia).21-23 A similarly expanded pool of bone calcium and increased calcium efflux from bone has been shown in dialysis patients with osteitis fibrosa studied with calcium isotopes.24 Moreover, in osteitis fibrosa, bone is deposited in a woven rather than the usual lamellar pattern. The abnormal woven bone may bind or incorporate calcium less well.25 Also, trabecular bone volume and thickness were greater in dialysis patients with high than low bone turnover suggesting that greater bone mass is present for calcium exchange in the former.25 A summary of the discussion on PTH and bone is provided in Table 1.
Table 1. Parathyroid hormone and calcium efflux from bone 
Phosphate
Phosphate is an important factor in evaluating the calcemic response to PTH. Long ago Albright showed that the first action of infused parathyroid extract was phosphaturia followed by lowering serum phosphorus before a calcemic effect developed.26 In the 1960s, Raisz showed in an ex-vivo study that increasing the phosphate concentration in the medium reduced calcium efflux from bone both in the absence and presence of PTH.27 Phosphate also modifies calcium efflux from bone in animal and human studies.28-30 In parathyroidectomized rats given a normal phosphate diet and a fixed replacement dose of PTH sufficient to maintain normal serum calcium and phosphorus values, changing to a high phosphate diet resulted in hypocalcemia and hyperphosphatemia.29 In young, healthy subjects given continuous intravenous phosphate infusion for 7 days, hypocalcemia and hyperphosphatemia developed until a three-fold increase in PTH increased phosphate excretion by five-fold and normalized serum calcium and phosphorus.28 Conversely, phosphate depletion induces hypercalcemia despite marked suppression of PTH.31 Even high dose bisphosphonate in phosphate-depleted rats did not prevent hypercalcemia despite producing toxic effects in osteoclasts and a marked decrease in the osteoblast surface.31 These results support the concept that an osteoclast independent effect may be important for bone efflux of calcium.32,33 Finally, bisphosphonate treatment in phosphate-depleted rats increased urine calcium excretion more than in phosphate-depleted rats not receiving bisphosphonates suggesting that decreased calcium influx to bone may be a factor.
In renal failure, many studies have shown that phosphate loading or hyperphosphatemia decreases the calcemic response to PTH.34-36 When 5/6 nephrectomy was performed in parathyroidectomized rats, a replacement dose of 1-34 PTH sufficient to maintain normal serum calcium and phosphorus values in parathyroidectomized rats with normal renal function failed to maintain normal serum calcium and phosphorus levels in renal failure.29 Even though dietary phosphate was unchanged, the phosphate burden was presumably increased because of decreased renal excretion of phosphate.29 Conversely, dietary phosphate restriction in renal failure decreases PTH while maintaining the serum calcium value. In essence, less PTH is needed to maintain the serum calcium concentration.37-40 Even in patients with advanced CKD, dietary phosphate restriction decreases PTH while maintaining normal serum calcium values.41,42 A summary of the discussion of the effect of phosphate on serum calcium and bone is provided in Table 2.
Table 2. Phosphorus and calcium efflux from bone 
Vitamin D
The effect of vitamin D on the exchangeable pool of bone calcium is more difficult to define than that of phosphate. Under some circumstances, 1,25 vitamin D (1,25D) appears to enhance, but in others to reduce calcium efflux from bone. In vitamin D intoxication, studies have suggested that calcium efflux from bone is increased.43-48 In vitro and in vivo studies have shown that calcitriol increases osteoclast activity.49-51 In a case report of vitamin D intoxication, bone histomorphometry showed an increase in osteoclasts with a reduction in osteoblasts, an increased osteoid surface and volume, and decreased mineralization.45 Studies in the chick also found that vitamin D intoxication impaired bone formation.52,53 The effects on osteoid volume and bone formation suggest that calcium influx to bone may be decreased in vitamin D intoxication. Both corticosteroids and bisphosphonates effectively treat hypercalcemia in vitamin D intoxication.43-45 However, in different settings, bisphosphonate treatment did not change the rate of recovery from EDTA-induced hypocalcemia in osteoporotic patients54 or prevent hypercalcemia in rats on a phosphate depleted diet perhaps suggesting different mechanisms.31
In a recent study in intestinal-specific VDR-null mice in which calcium absorption is unresponsive to 1,25D, insights into the calcemic and bone effects of 1,25D during calcium deficiency were provided.55 In this mouse model, markedly elevated endogenous 1,25D levels maintained serum calcium normal by increasing calcium efflux from bone while bone mineralization was inhibited resulting in hyperosteoidosis and a decrease in bone mass. The activation of bone resorption by 1,25D in calcium deficient intestinal-specific VDR-null mice was osteoblast mediated. In systemic VDR-receptor null mice, bone abnormalities were prevented when calcium repletion was achieved with a high calcium/lactose diet.56 In the intestinal-specific null model, although PTH values were elevated, the authors concluded that because markedly elevated PTH did not prevent hypocalcemia in systemic VDR null mice, 1,25D played a critical role in increasing calcium efflux from bone in intestinal-specific VDR-null mice. Finally, the calcemic bone actions of 1,25D were blocked by bisphosphonates. The authors concluded that high 1,25D levels in calcium deficiency: 1) increase calcium efflux from bone; 2) decrease calcium influx to bone by inhibiting bone mineralization; and 3) both 1) and 2) act to maintain serum calcium, but decrease bone mass.
In intestinal-specific VDR null mice, a marked elevation in 1,25D increased calcium efflux from bone.55 But a major question is whether such a mechanism is applicable in vitamin D deficiency? In dogs placed on a vitamin D and calcium deficient diet, 25D values decreased rapidly, but 1,25D values tripled by 24 weeks and remained greater than normal at 36 weeks before subsequently declining below normal.57 Ionized calcium values remained normal until 24 weeks before marginally decreasing at 36 weeks. Thus, despite vitamin D and calcium deficiency, serum calcium values were maintained normal for a prolonged time as 1,25D and PTH values tripled during the first 24 weeks. An elevated 1,25D and PTH together with hypophosphatemia contributed to the prolonged maintenance of serum calcium. The marked 1,25D increase during the first 24 weeks could have helped maintain serum calcium by enhancing calcium efflux from bone while also decreasing calcium influx to bone. In humans with established vitamin D deficiency, studies have shown that treatment with vitamin D or 25D increases 1,25D values to 3 to 4 times greater than normal.58-60 Thus, it is plausible in vitamin D deficiency that high 1,25D values have an important skeletal effect in maintaining serum calcium during its evolution and increasing serum calcium during initial treatment with vitamin D. Also, in evolving vitamin D deficiency, high 1,25D values perhaps together with PTH-induced decreases in serum phosphorus potentially contribute to the development of osteomalacia.
In renal failure, several studies have shown that acute calcitriol administration increases the calcemic response to PTH.61,62 In one study, high dose PTH was infused for 48 hours while calcitriol was administered to rats on a calcium free diet.20 Thus, neither calcitriol nor hypercalcemia could modify PTH levels. Calcitriol administration enhanced the calcemic response to PTH in azotemic rats on a low phosphate diet and in both normal and azotemic rats on a high phosphate diet (Figure 2 B).20
Two studies in dialysis patients are of particular interest because long-term calcitriol treatment decreased the bone response to PTH.63,64 In one study, 14 children on peritoneal dialysis with biopsy-proven osteitis fibrosa were treated with high dose calcitriol for 12 months with dose adjustments for hypercalcemia and hyperphosphatemia.63 The average calcitriol dose was 1.25μg thrice weekly. The dialysate calcium concentration was 3.5meq/L and calcium carbonate was the primary phosphate binder. A repeat bone biopsy performed after one year showed that 12 patients developed normal or adynamic bone and in 6 patients the bone formation rate decreased despite no change in PTH values. The latter result suggests that calcitriol treatment may have decreased the bone formation rate similar to results seen in intestinal-specific VDR null mice.55 A positive calcium balance from calcium carbonate and high dialysate calcium concentration could also have contributed.
In the other study, a hemodialysis patient with markedly elevated intact PTH (1912pg/mL, Nichols assay) and serum alkaline phosphatase (1512IU/L) was studied before calcitriol treatment, after 6 weeks of 2μg intravenous calcitriol thrice weekly, and 6 weeks after calcitriol was stopped.64 Studies consisted of separate dialysis sessions with low (1meq/L) and high (4meq/L) calcium dialysate to determine maximal PTH secretion and suppression respectively. Despite 6 weeks of calcitriol treatment, predialysis serum calcium and PTH values were similar to pretreatment values (Figure 3 A and Figure 3 B) as were serum phosphorus and alkaline phosphatase values. The maximal PTH value during induction of hypocalcemia was similar approaching 4000pg/mL during both low calcium dialysis studies (Figure 3 B). However, during the low calcium dialysis in the first study, serum calcium, which started at 10.8mg/dL, only decreased to 9.4mg/dL at 180 minutes. After calcitriol treatment, despite a similar starting serum calcium concentration, serum calcium decreased to less than 8mg/dL after only 15 minutes of dialysis (Figure 3A). Studies with the high calcium dialysate showed marked differences in bone buffering of calcium. Before calcitriol treatment, serum calcium slowly increased to 11.8mg/dL at 180 minutes. However, after calcitriol treatment, serum calcium increased to 12.2mg/dL after only 15 minutes (Figure 3 C). PTH suppression was similar (Figure 3 D). When dialysis studies were repeated after calcitriol had been stopped for 6 weeks, results were similar to those before calcitriol was started (Figure 3). Thus, in this hemodialysis patient with severe secondary hyperparathyroidism and an enhanced capacity to defend against the induction of hypocalcemia and hypercalcemia, calcitriol treatment for 6 weeks dramatically decreased calcium efflux from and influx to bone.

In both cited studies, a decreased bone/calcemic response to PTH was seen. In the second study, the exchangeable pool of bone calcium was seemingly greatly reduced by calcitriol treatment despite no change in PTH secretion. In the first study it could be argued that the decrease in osteoblasts and osteoclasts, which reflects bone remodeling, is different from an evaluation of bone efflux and influx of calcium. However, the marked reduction in bone formation rate together with the decreased cellular activity seen after calcitriol treatment is also a likely indicator of a decreased exchangeable pool of bone calcium.
How does the vitamin D/calcium biochemical profile differ between dialysis patients receiving high doses of calcitriol and patients with vitamin D intoxication? In dialysis patients, calcitriol treatment reduced calcium efflux from bone during a low calcium dialysis while in patients with vitamin D intoxication increased calcium efflux from bone is reported to be a major contributor to hypercalcemia. In vitamin D intoxication, hypercalcemia is persistent with suppressed PTH. In dialysis patients treated with calcitriol, if monitored diligently the serum calcium concentration is normal while the high PTH value generally decreases, but remains elevated. In the study in which calcitriol was given thrice weekly, measured serum calcitriol values were in the normal range before subsequent dosing,63 but high 1,25D values would be expected immediately after calcitriol dosing.65 In dialysis patients, 25OHD values are often less than the desired goal, 30ng/mL. Conversely, in vitamin D intoxication, 25OHD values are markedly elevated while serum calcitriol values are often but not always increased.66-68
How does vitamin D affect calcium efflux from bone in vitamin D intoxication? In vitamin D intoxication, longstanding hypercalcemia leads to reduced renal function further exacerbating hypercalcemia, which may act to drive calcium into bone. Treatment with normal saline improves renal function by correcting the decreased extracellular volume, which in turn, increases renal excretion of calcium lowering serum calcium. As a result, the supersaturated bone releases more calcium for several reasons.43-45 These include an increasing calcium gradient between bone and blood, reduced serum phosphorus from improved renal function, and increased PTH secretion from a reduction in serum calcium. Such actions could account for the enhanced calcium release from bone reported in vitamin D intoxication. Finally, suggesting that increased bone calcium deposition may occur during the hypercalcemia of vitamin D intoxication is the report of increased bone mineral density in osteoporotic patients with vitamin D intoxication.48
Why did high dose calcitriol treatment decrease the exchangeable pool of bone calcium in the dialysis patient with severe secondary hyperparathyroidism? In dialysis patients with osteitis fibrosa, there are abundant osteoblasts in which the VDR can be activated to generate osteoclasts and increase bone resorption. However, calcitriol administration decreased the exchangeable pool of bone calcium. In both cited studies in patients with osteitis fibrosa, a calcium-replete environment was present due to high dialysate calcium, the use of calcium-based phosphate binders, and intermittent or continual hypercalcemia.63,64 In contrast, in studies in intestinal-specific VDR-null mice in which 1,25D-mediated increased bone resorption and decreased bone formation were seen, calcium depletion was present.55 However, when the VDR-null mouse was calcium replete, increased bone resorption and decreased bone formation were prevented.56 Other important differences are present between dialysis patients with osteitis fibrosa and intestinal-specific VDR null mice/patients with vitamin D deficiency. In the dialysis patients, calcitriol administration was intermittent in contrast to continuously elevated, endogenous 1,25D values. Although PTH values are increased in all the cited conditions, the calcium and phosphorus status are quite different. In the dialysis patients, hypercalcemia and hyperphosphatemia are present while in intestinal specific null mice and vitamin D deficient patients, hypocalcemia and hypophosphatemia prevail. In the reported dialysis patient,64 increases in serum calcium were not seen with calcitriol treatment. However, whether increased bone buffering of calcium not only prevented hypercalcemia, but also drove the mineralization process decreasing the exchangeable pool of bone calcium is an interesting possibility. A summary of the discussion on 1,25D and bone is provided Table 3.
Table 3. Calcitriol and calcium efflux from bone 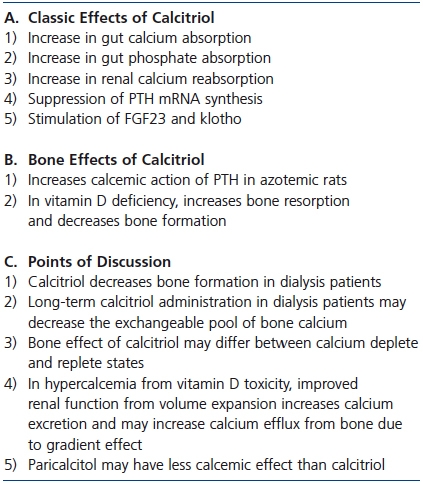
A final point of discussion is the effect of both 19-nor-1,25-dihydroxy-vitamin D2 (paricalcitol), an analog of 1,25D, and the calcimimetic, cinacalcet, on bone resorption and serum calcium. Both agents have been used for the past 10 to 15 years for the treatment of hyperparathyroidism in dialysis patients. To achieve similar PTH suppression, the dose ratio of paricalcitol to calcitriol used has been 3 or 4:1. Most, but not all in vitro and animal studies have shown that the calcemic effect of paricalcitol is less than that of calcitriol.69-71 In studies in dialysis patients, paricalcitol suppresses PTH secretion at least as well if not better than calcitriol72 and may also have less calcemic effect.72,73 In studies in uremic rats, less vascular calcification has been seen with paricalcitol than with calcitriol74 and calcimimetics appear to be even more effective in preventing vascular calcification.75 A summary of the discussion on 1,25D and bone is provided Table 3.
Calcimimetics are an effective treatment for hyperparathyroidism in the uremic animals and dialysis patient. Because of its calcimimetic activity, hypocalcemia generally develops during treatment. Several studies in rats have evaluated the effect of calcimimetics on bone.76-78 Two studies in rats used a 5/6 nephrectomy model76,78 and the other an adenine model of uremia.77 The latter model results in much more severe renal failure, hyperparathyroidism, hypocalcemia and hyperphosphatemia than the former model. In one of the 5/6 nephrectomy models, a high phosphate diet was used78 and in the other a relatively high calcium diet was used.76 Moreover, different calcimimetics were used in the three studies. In all the studies, serum calcium and PTH decreased, but the magnitude of the reduction in PTH differed. Changes in bone histology also differed considerably among the studies. In another study simulating primary hyperparathyroidism, the bone response to a two week continuous infusion of PTH was evaluated in mice with either a PTH gene knockout or PTH and calcium sensing receptor gene knockout (double knockout). The mice had normal renal function. The absence of the calcium sensing receptor reduced the bone effects of PTH in this model of primary hyperparathyroidism.79
Uremic toxins
A variety of factors which could potentially contribute to skeletal resistance to PTH are altered in CKD. Exposure of osteoblast-like cells to uremic serum affects the number and function of these cells including suppressed mitogenesis and IL-6 release and reduced PTHR expression.80-82 In CKD, levels of inhibitory and stimulatory insulin growth factor binding proteins 4 and 5 respectively, are altered and may contribute to abnormal osteoblast proliferation.80 Osteoprotegerin, a decoy receptor of NF-kB ligand (RANKL), accumulates as renal failure progresses blocking osteoclast activation by PTH.
The concentration of absorbed products of intestinal microbial flora increase as renal function declines. These products, particularly indoxyl sulfate and p-cresol sulfate, may alter bone metabolism and contribute to bone resistance to PTH. Indole, formed by gastrointestinal bacteria during breakdown of tryptophan, is metabolized to indoxyl sulfate which is transported by an organic anion transporter into osteoblasts where it induces oxidative stress. Exposure of osteoblast cells in culture to indoxyl sulfate inhibits PTH-induced cyclic AMP generation and downregulates PTHR expression, an effect partially inhibited by the organic anion transporter inhibitor, probenicid.83 Elevated indoxyl sulfate values in hemodialysis patients inversely correlate with markers of bone formation.84 Furthermore, in a rat model of adynamic bone in which elevated indoxyl sulfate values were present, administration of the oral absorbent, AST-120, lowered indoxyl sulfate values and increased bone formation.85
Indoxyl sulfate and p-cresol sulfate, a gut microbial product produced from metabolism of tyrosine, may indirectly alter PTH action on bone through the FGF23-klotho axis. Klotho blood values are low in CKD while FGF23 values are markedly elevated. In a mouse model of CKD and in isolated kidney cells, both indoxyl sulfate and p-cresol sulfate suppressed klotho production by epigenetic modification of the klotho gene.86 These toxins stimulate expression of DNA methyltransferase 1 which hypermethylates the klotho gene suppressing gene expression. A deficiency of klotho as exemplified by the klotho knockout mouse results in elevated values of serum calcium, phosphorus and 1,25D, ectopic calcification including extensive vascular calcification, and osteoporosis with decreases in osteoblasts and osteoclasts.87 FGF23 values are markedly elevated in the klotho knockout model similar to that in CKD. Thus, reduced levels of klotho in CKD, perhaps at least in part due to uremic toxins, could alter the bone response to PTH via hyperphosphatemia and/or extremely elevated FGF23 levels found in CKD.88 The potential role of uremic toxins on bone is summarized in Table 4.
Table 4. Uremic toxins and calcium efflux from bone 
A summary of the many factors affecting calcium efflux from bone discussed in our review is shown in Figure 4. Many unanswered questions exist about the exchangeable pool of bone calcium. These include 1) how PTH mobilizes calcium from bone, 2) what is the contribution of osteoclast-mediated calcium efflux in contrast to shifts across the quiescent bone surface, 3) how after induced hypocalcemia, serum calcium is restored to baseline values in parathyroidectomized animals, 4) how parathyroidectomy restores the calcemic response to PTH to normal in the azotemic rat, 5) how phosphate modifies calcium efflux from bone, 6) why bisphosphonate treatment reduces calcium efflux from bone in vitamin D intoxication, but not in phosphate depletion, 7) how calcitriol treatment decreases the exchangeable pool of bone calcium in dialysis patients with osteitis fibrosa, and 8) the role of uremic toxins on the exchangeable pool of bone calcium. Much remains to be learned about the role of bone in the regulation of serum calcium.

Disclosures
B.L and A.F. have no disclosures. M.R. has received grants from Amgen and Fresenius and lecture fees from Amgen, Abbott, Shire and Fresenius.
Conflicts of interest
The authors declare that they have no conflicts of interest related to the contents of this article.
Referencias Bibliográficas
1. Massry SG, Coburn JW, Lee DBW, Jowsey J, Kleeman CR. Skeletal resistance to parathyroid hormone in renal failure. Studies in 105 human subjects. Ann Intern Med 1973;78:357-64. [ Links ]
2. Wesseling-Perry K, Harkins GC, Wang HJ, Elashoff R, Gales B, Horwitz MJ, et al. The calcemic response to continuous parathyroid hormone (PTH)(1-34) infusion in end-stage kidney disease varies according to bone turnover: A potential role for PTH (7-84). J Clin Endocrinol Metab 2010;95:2772-80. [ Links ]
3. Brent GA, LeBoff MS, Seely EW, Conlin PR, Brown EM. Relationship between the concentration and rate of change of calcium and serum intact parathyroid hormone levels in normal humans. J Clin Endocrinol Metab 1988;67:944-50. [ Links ]
4. Berdud I, Martin Malo A, Almaden Y, Aljama P, Rodriguez M, Felsenfeld AJ. The PTH-calcium relationship during a range of infused PTH doses in the parathyroidectomized rat. Calcif Tissue Int 1998;62:457-61. [ Links ]
5. Wang W, Lewin E, Olgaard K. PTH is not a key hormone in the rapid minute-to-minute regulation of the plasma Ca homeostasis in the rat. Eur J Clin Invest 1999;29:309-20. [ Links ]
6. Alexander RL Jr. Calcium homeostasis in the thyroparathyroidectomized dog. Endocrinology 1965;77:985-90. [ Links ]
7. Felsenfeld AJ. Bone, parathyroid hormone and the response to the rapid induction of hypocalcemia. Eur J Clin Invest 1999;29:274-7. [ Links ]
8. Nguyen-Yamamoto L, Rousseau L, Brossard JH, Lepage R, D'Amour P. Synthetic carboxyl-terminal fragments of parathyroid hormone (PTH) decrease ionized calcium concentrations in rats by acting on a receptor different from the PTH/PTH-related peptide receptor. Endocrinology 2001;142:1386-92. [ Links ]
9. Parfitt AM. Calcium homeostasis. In: Mundy GR, Martin TJ (eds.). Physiology and pharmacology of bone. Chap 1. Heidelberg: Springer-Verlag; 1993. pp.1-65. [ Links ]
10. Talmage RV. Effect of fasting and parathyroid hormone injection on plasma 45Ca concentrations in rats. Calcif Tissue Res 1975;17:103-12. [ Links ]
11. Wong KM, Klein L. Circadian variations in contributions of bone and intestine to plasma calcium in dogs. Am J Physiol 1984;246:R688-92. [ Links ]
12. D'Amour P, Palardy J, Bahsali G, Mallette LE, DeLéan A, Lepage R. The modulation of circulating parathyroid hormone immunoheterogeneity in man by ionized calcium concentration. J Clin Endocrinol Metab 1992;74:525-32. [ Links ]
13. Brossard JH, Cloutier M, Roy L, Lepage R, Gascon-Barré M, D'Amour P. Accumulation of a non-(1-84) molecular form of parathyroid hormone (PTH) detected by intact PTH assay in renal failure: Importance in the interpretation of PTH values. J Clin Endocrinol Metab 1996;81:3923-9. [ Links ]
14. Slatopolsky E, Finch J, Clay P, Martin D, Sicard G, Singer G, et al. A novel mechanism for skeletal resistance in uremia. Kidney Int 2000;58:753-61. [ Links ]
15. Divieti P, John MR, Juppner H, Bringhurst FR. Human PTH-(7-84) inhibits bone resorption in vitro via actions independent of the type 1 PTH/PTHrP receptor. Endocrinology 2002;143:171-6. [ Links ]
16. Murray TM, Rao LG, Divieti P, Bringhurst FR. Parathyroid hormone secretion and action: Evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxy-terminal ligands. Endocr Rev 2005;26:78-113. [ Links ]
17. Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab 2010;21:369-74. [ Links ]
18. Friedman PA, Goodman WG. PTH(1-84)/PTH(7-84): a balance of power. Am J Physiol Renal Physiol 2006;290:F975-84. [ Links ]
19. Usatii M, Rousseau L, Demers C, Petit JL, Brossard JH, Gascon-Barré M, et al. Parathyroid hormone fragments inhibit active hormone and hypocalcemia-induced 1,25(OH)2D synthesis. Kidney Int 2007;72:1330-5. [ Links ]
20. Rodriguez M, Felsenfeld AJ, Llach F. Calcemic response to parathyroid hormone in renal failure: Role of calcitriol and the effect of parathyroidectomy. Kidney Int 1991;40:1063-8. [ Links ]
21. Andress D, Felsenfeld AJ, Voigts A, Llach F. Parathyroid hormone response to hypocalcemia in hemodialysis patients with osteomalacia. Kidney Int 1983;24:364-70. [ Links ]
22. Voigts A, Felsenfeld AJ, Andress D, Llach F. Parathyroid hormone and bone histology: Response to hypocalcemia in osteitis fibrosa. Kidney Int 1984;25:445-52. [ Links ]
23. Felsenfeld AJ, Rodriguez M, Dunlay R, Llach F. A comparison of parathyroid-gland function in haemodialysis patients with different forms of renal osteodystrophy. Nephrol Dial Transplant 1991;6:244-51. [ Links ]
24. Kurz P, Monier-Faugere MC, Bognar B, Werner E, Roth P, Vlachojannis J, et al. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int 1994;46:855-61. [ Links ]
25. Malluche HH, Porter DS, Monier-Faugere MC, Mawad H, Pienkowski D. Differences in bone quality in low- and high-turnover renal osteodystrophy. J Am Soc Nephrol 2012;23:525-32. [ Links ]
26. Albright F, Bauer W, Ropes M, Aub JC. Studies of calcium and phosphorus metabolism. IV. The effect of the parathyroid hormone. J Clin Invest 1929;7:139-81. [ Links ]
27. Raisz LG, Niemann I. Effect of phosphate, calcium, and magnesium on bone resorption and hormonal responses in tissue culture. Endocrinology 1969;85:446-52. [ Links ]
28. Krapf R, Glatz M, Hulter HN. Neutral phosphate administration generates and maintains renal metabolic alkalosis and hyperparathyroidism. Am J Physiol 1995;268:F802-7. [ Links ]
29. Tallon S, Berdud I, Hernandez A, Concepcion MT, Almaden Y, Torres A, et al. The relative effects of PTH and dietary phosphorus on calcitriol production in normal and azotemic rats. Kidney Int 1996;49:1441-6. [ Links ]
30. Li F, Muhlbauer RC. Food fractionation is a powerful tool to increase bone mass in growing rats and to decrease bone mass in aged rats: Modulation of the effect of dietary phosphate. J Bone Miner Res 1999;14:1457-65. [ Links ]
31. Jara A, Lee E, Stauber D, Moatamed F, Felsenfeld AJ, Kleeman CR. Phosphate depletion in the rat: effect of bisphosphonates and the calcemic response to PTH. Kidney Int 1999;55:1434-43. [ Links ]
32. Parfitt AM. Misconceptions (3): calcium leaves bone only by resorption and enters only by formation. Bone 2003;33:259-63. [ Links ]
33. Marenzana M, Shipley AM, Squitiero P, Kunkel JG, Rubinacci A. Bone as an ion exchange organ: evidence for instantaneous cell-dependent calcium efflux from bone not due to resorption. Bone 2005;37:545-54. [ Links ]
34. Somerville PJ, Kaye M. Evidence that resistance to the calcemic action of parathyroid hormone in rats with acute uremia is caused by phosphate retention. Kidney Int 1979;16:552-60. [ Links ]
35. Somerville PJ, Kaye M. Action of phosphorus on calcium release in isolated perfused rat tails. Kidney Int 1982;22:348-54. [ Links ]
36. Rodriguez M, Martin Malo A, Martinez ME, Torres A, Felsenfeld AJ, Llach F. Calcemic response to parathyroid hormone in renal failure: role of phosphorus and its effect on calcitriol. Kidney Int 1991;40:1055-62. [ Links ]
37. Slatopolsky E, Caglar S, Gradowska L, Canterbury J, Reiss E, Bricker NS. On the prevention of secondary hyperparathyroidism in experimental chronic renal disease using "proportional reduction" of dietary phosphorus intake. Kidney Int 1972;2:147-51. [ Links ]
38. Kaplan MA, Canterbury JM, Bourgoignie JJ, Veliz G, Gavellas G, Reiss E, et al. Reversal of hyperparathyroidism in response to dietary phosphorus restriction in uremic dogs. Kidney Int 1979;15:43-8. [ Links ]
39. Portale AA, Booth BE, Halloran B, Morris RC Jr. Effect of dietary phosphorus on circulating concentrations of 1,25-dihydroxyvitamin D and immunoreactive parathyroid hormone in children with moderate renal insufficiency. J Clin Invest 1984;73:1580-9. [ Links ]
40. Lopez-Hilker S, Dusso AS, Rapp NS, Martin KJ, Slatopolsky E. Phosphorus restriction reverses secondary hyperparathyroidism independent of changes in calcium and calcitriol. Am J Physiol 1990;259:F432-7. [ Links ]
41. Combe C, Aparicio M. Phosphorus and protein restriction and parathyroid function in chronic renal failure. Kidney Int 1994;46:1381-6. [ Links ]
42. Combe C, Morel D, de Precigout V, Blanchetier V, Bouchet JL, Potaux L, et al. Long-term control of hyperparathyroidism in advanced renal failure by low-phosphorus diet supplemented with calcium (without changes in plasma calcitriol). Nephron 1995;70:287-95. [ Links ]
43. Streck W, Waterhouse C, Haddad JG. Glucocorticoid effects in vitamin D intoxication. Arch Intern Med 1979;139:974-7. [ Links ]
44. Heyburn PJ, Francis RM, Peacock M. Acute effects of saline, calcitonin, and hydrocortisone on plasma calcium in vitamin D intoxication. Br Med J 1979;1:232-3. [ Links ]
45. Davies M, Mawer EB, Freemont AJ. The osteodystrophy of hypervitaminosis D: A metabolic study. Q J Med 1986;61:911-9. [ Links ]
46. Rizzoli R, Stoermann C, Ammann P, Bonjour JP. Hypercalcemia and hyperosteolysis in vitamin D intoxication: effects of clodronate therapy. Bone 1994;15:193-8. [ Links ]
47. Selby PL, Davies M, Marks JS, Mawer EB. Vitamin D intoxication causes hypercalcaemia by increased bone resorption which responds to pamidronate. Clin Endocrinol (Oxf) 1995;43:531-6. [ Links ]
48. Adams JS, Lee G. Gains in bone mineral density with resolution of vitamin D intoxication. Ann Intern Med 1997;127:203-6. [ Links ]
49. Raisz LG, Trummel CL, Holick MF, DeLuca HF. 1,25-Dihydroxycholecalciferol: a potent stimulator of bone resorption in tissue culture. Science 1972;175:768-9. [ Links ]
50. Holtrop ME, Cox KA, Clark MB, Holick MF, Anast CS. 1,25-dihydroxycholecalciferol stimulates osteoclasts in rat bones in the absence of parathyroid hormone. Endocrinology 1981;108:2293-301. [ Links ]
51. Miao D, He B, Lanske B, Bai XY, Tong XK, Hendy GN, et al. Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology 2004;145:2046-53. [ Links ]
52. Chen PS Jr., Bosmann B. Comparison of the hypercalcemic action of vitamins D2 and D3 in chicks and the effect on tetracycline fixation by bone. J Nutr 1965;87:148-54. [ Links ]
53. Klein L. Direct measurement of bone resorption and calcium conservation during vitamin D deficiency or hypervitaminosis D. Proc Natl Acad Sci U S A 1980;77:1818-22. [ Links ]
54. Landman JO, Schweitzer DH, Frolich M, Hamdy NA, Papapoulos SE. Recovery of serum calcium concentrations following acute hypocalcemia in patients with osteoporosis on long-term oral therapy with the bisphosphonate pamidronate. J Clin Endocrinol Metab 1995;80:524-8. [ Links ]
55. Lieben L, Masuyama R, Torrekens S, Van Looveren R, Schrooten J, Baatsen P, et al. Normocalcemia is maintained in mice under conditions of calcium by vitamin D-induced inhibition of bone mineralization. J Clin Invest 2012;122:1803-15. [ Links ]
56. Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, et al. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology 1999;140:4982-7. [ Links ]
57. Cloutier M, Gascon-Barre M, D'Amour P. Chronic adaptation of dog parathyroid function to a low-calcium-high-sodium-Vitamin D-deficient diet. J Bone Miner Res 1992;7:1021-8. [ Links ]
58. Papapoulos SE, Clemens TL, Fraher LJ, Gleed J, O'Riordan JL. Metabolites of vitamin D in human vitamin-D deficiency: effect of vitamin D3 or 1,25-dihydroxy-cholecalciferol. Lancet 1980;2:612-5. [ Links ]
59. Stanbury SW, Taylor CM, Lumb GA, et al. Formation of vitamin D metabolites following correction of human vitamin D deficiency. Miner Electrolyte Metab 1981;5:212-27. [ Links ]
60. Venkataraman PS, Tsang RC, Buckley DD, Ho M, Steichen JJ. Elevation of serum 1,25-dihydroxyvitamin D in response to physiologic doses of vitamin D in vitamin D-deficient infants. J Pediatr 1983;103:416-9. [ Links ]
61. Massry SG, Stein R, Garty J, Arieff AI, Coburn JW, Norman AW, et al. Skeletal resistance to the calcemic action of parathyroid hormone in uremia: Role of 1,25(OH)2D3. Kidney Int 1976;9:467-74. [ Links ]
62. Massry SG, Tuma S, Dua A, Goldstein DA. Reversal of skeletal resistance to parathyroid hormone in uremia by vitamin D metabolites. Evidence for requirement of 1,25(OH)2D3. J Lab Clin Med 1979;94:152-7. [ Links ]
63. Goodman WG, Ramirez JA, Belin TR, Chon Y, Gales B, Segre GV, et al. Development of adynamic bone in patients with secondary hyperparathyroidism after intermittent calcitriol therapy. Kidney Int 1994;46:1160-6. [ Links ]
64. Pahl M, Jara A, Bover J, Felsenfeld AJ. Studies in a hemodialysis patient indicating that calcitriol may have a direct suppressive effect on bone. Nephron 1995;71:218-23. [ Links ]
65. Levine BS, Song M. Pharmacokinetics and efficacy of pulse oral versus intravenous calcitriol in hemodialysis patients. J Am Soc Nephrol 1996;7:488-96. [ Links ]
66. Araki T, Holick MF, Alfonso BD, Charlap E, Romero CM, Rizk D, et al. Vitamin D intoxication with severe hypercalcemia due to manufacturing and labeling errors of two dietary supplements made in the United States. J Clin Endocrinol Metab 2011;96:3603-8. [ Links ]
67. Lowe H, Cusano NE, Binkley N, Blaner WS, Bilezikian JP. Vitamin D toxicity due to a commonly available "over the counter" remedy from the Dominican Republic. J Clin Endocrinol Metab 2011;96:291-5. [ Links ]
68. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr 2008;88:582S-586S. [ Links ]
69. Finch JL, Brown AJ, Slatopolsky E. Differential effects of 1,25-dihydroxyvitamin D3 and 19-nor-1,25-dihydroxyvitamin D2 on calcium and phosphorus resorption in bone. J Am Soc Nephrol 1999;10:980-5. [ Links ]
70. Balint E, Marshall CF, Sprague SM. Effect of vitamin D analogues paracalcitol and calcitriol on bone mineral in vitro. Am J Kidney Dis 2000;36:789-96. [ Links ]
71. Nakane M, Fey TA, Dixon DB, Ma J, Brune ME, Li YC, et al. Differential effects of vitamin D analogs on bone formation and resorption. J Steroid Biochem Mol Biol 2006;98:72-7. [ Links ]
72. Sprague SM, Llach F, Amdahl M, Taccetta C, Batlle D. Paracalcitol versus calcitriol in the treatment of secondary hyperparathyroidism. Kidney Int 2003;63:1483-90. [ Links ]
73. Coyne DW, Grieff M, Ahya SN, Giles K, Norwood K, Slatopolsky E. Differential effects of acute administration of 19-nor-1,25-dihydroxy-vitamin D2 and 1,25-dihydroxy-vitamin D3 on serum calcium and phosphorus in hemodialysis patients. Am J Kidney Dis 2002;40:1283-8. [ Links ]
74. Cardus A, Panizo S, Parisi E, Fernandez E, Valdivielso JM. Differential effects of vitamin D analogs on vascular calcification. J Bone Miner Res 2007;22:860-6. [ Links ]
75. Rodriguez M, Aguilera-Tejero E, Mendoza FJ, Guerrero F, López I. Effects of calcimimetics on extraskeletal calcifications in chronic kidney disease. Kidney Int Suppl 2008;(111):S50-4. [ Links ]
76. Wada M, Ishii H, Furuya Y, Fox J, Nemeth EF, Nagano N. NPS R-568 halts or reverses osteitis fibrosa in uremic rats. Kidney Int 1998;53:448-53. [ Links ]
77. Henley C, Davis J, Miller G, Shatzen E, Cattley R, Li X, et al. The calcimimetic AMG 641 abrogates parathyroid hyperplasia, bone and vascular calcification abnormalities in uremic rats. Eur J Pharmacol 2009;616:306-13. [ Links ]
78. Finch JL, Tokumoto M, Nakamura H, Yao W, Shahnazari M, Lane N, et al. Effect of paracalcitol and cinacalcet on serum phosphate, FGF-23, and bone in rats with chronic kidney disease. Am J Physiol Renal Physiol 2010;298:F1315-22. [ Links ]
79. Xue Y, Xiao Y, Liu J, Karaplis AC, Pollak MR, Brown EM, et al. The calcium-sensing receptor complements parathyroid hormone-induced bone turnover in discrete skeletal compartments in mice. Am J Physiol Endocrinol Metab 2012;302:E841-51. [ Links ]
80. Wagner MS, Stracke S, Jehle PM, Keller F, Zellner D, Baylink DJ, et al. Evaluation of IGF system component levels and mitogenic activity of uremic serum on normal human osteoblasts. Nephron 2000;84:158-66. [ Links ]
81. Disthabanchong S, Hassan H, McConkey CL, Martin KJ, Gonzalez EA. Regulation of PTH1 receptor expression by uremic ultrafiltrate in UMR 106-01 osteoblast-like cells. Kidney Int 2004;65:897-903. [ Links ]
82. Steddon SJ, McIntyre CW, Schroeder NJ, Burrin JM, Cunningham J. Impaired release of interleukin-6 from human osteoblastic cells in the uraemic mileau. Nephrol Dial Transplant 2004;19:3078-83. [ Links ]
83. Iwasaki Y, Yamato H, Nii-Kono T, Fujieda A, Uchida M, Hosokawa A, et al. Uremic toxin and bone metabolism. J Bone Miner Metab 2006;24:172-5. [ Links ]
84. Goto S, Fujii H, Hamada Y, Yoshiya K, Fukagawa M. Association between indoxyl sulfate and skeletal resistance in hemodialysis patients. Ther Apher Dial 2010;14:417-23. [ Links ]
85. Iwasaki Y, Yamato H, Nii-Kono T, Fujieda A, Uchida M, Hosokawa A, et al. Administration of oral charcoal adsorbent (AST-120) suppresses low-turnover bone progression in uraemic rats. Nephrol Dial Transplant 2006;21:2768-74. [ Links ]
86. Sun CY, Chang SC, Wu MS. Suppression of klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int 2012;81:640-50. [ Links ]
87. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the klotho gene leads to a syndrome resembling ageing. Nature 1997;390:45-51. [ Links ]
88. Feng JQ, Ye L, Schiavi S. Do osteocytes contribute to phosphate homeostasis? Curr Opin Nephrol Hypertens 2009;18:285-91. [ Links ]
![]() Correspondence:
Correspondence:
Arnold J. Felsenfeld
Departments of Medicine
VA Greater Los Angeles Healthcare System and
the David Geffen School of Medicine at UCLA
Los Angeles, CA, USA
Arnold.Felsenfeld@va.gov
Enviado a Revisar: 28 Nov. 2013
Aceptado el: 25 Jun. 2014

