










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkDear Editor,
Atypical hemolytic-uremic syndrome (aHUS) is an extremely rare, genetic, chronic, and progressive inflammatory disease1 caused by defects in complement system. These defects result in systemic thrombotic microangiopathy involving damage to multiple organ systems including renal dysfunction.1-7 Genetic mutations have been detected in around 50% of the reported cases. Regarding renal transplant patients, expanded criteria donors, infection by cytomegalovirus or BK, use of CNI or m-TOR inhibitors and antibody-mediated rejection (AMR) have been related to de novo post-transplant aHUS.8 Graft failure is reported in 60-90% of patients within 1 year.10 Eculizumab is a monoclonal antibody that binds to C5 complement protein avoiding the formation of the cell membrane attack complex.6,7 We report herein a case of late onset of de novo post-transplant aHUS on a simultaneous pancreas and kidney recipient with severe systemic manifestations, without presenting acute graft rejection, successfully treated with limited doses of eculizumab remaining stable after one year of follow-up.
A 46-year-old woman with end stage renal disease secondary to type 1 Diabetes Mellitus underwent simultaneous pancreas and kidney transplant on October 2012 from deceased donor. Received induction with Basiliximab and maintenance treatment with mycophenolate mofetil, tacrolimus, and prednisone. Pancreatic and renal function were stable, creatinine (SCr) of 110 μmol/L. Tacrolimus through levels remained between 6 and 8 ng/ml. Seven months after transplant presented fever, abdominal pain, diarrhea and vomiting, acute graft dysfunction (SCr 438 μmol/L) and thrombocytopenia (53 × 10 × 9/L). She developed progressive anemia and thrombocytopenia and worsening of renal function requiring dialysis. Pancreas graft function remained preserved. Thrombotyc microangiopathy (TMA)was detected with lactate dehydrogenase (LDH) up to 1500 UI/L, undetectable haptoglobin and schistocytes. C3 was low, ADAMTS 13 of 79% and Shigella toxin was negative. Urinary tract infection by extended-spectrum beta-lactamase (ESBL)-klebsiella was present. Initial biopsy confirmed findings of TMA with severe tubulo-interstitial involvement and some focal glomerular involvement, without features of acute rejection. Discontinuation of tacrolimus, antibiotic treatment with meropenem and daily plasma exchange (PE) for 10 days were performed. The patient did not respond to therapy. Second renal biopsy showed persistent signs of TMA, worsening involvement of glomeruli with mesangiolysis without rejection (Fig. 1). Treatment with Eculizumab was started with 4 doses of 900 mg Eculizumab iv on a weekly basis, showing improvement of renal function, cessation of hemodialysis and hemolysis. Genetic screening did not detect mutations on factor I, factor H or factor MCP genes. Risk haplotype in heterozygosis for factor H and MCP genes were observed. Study of the complement alternate pathway showed low factor C3, factor H, normal expression of MCP (membrane cofactor protein) and negative anti-factor H antibodies. A third biopsy was performed showing predominance of chronic lessions of TMA and mild signs of acute TMA with interstitial fibrosis of 5-10% and some microhemorrhages, with ATN (Fig. 2). New course of 3 daily PE and final dose of 1200 mg of Eculizumab iv were prescribed. She presented normalized C3, LDH, haptoglobin, hemoglobin and platelets. At discharge Scr was 300 μmol/L. Immunosupresive maintenance therapy at discharge was prednisone and mycophenolate mofetil. Renal function continued to improve and at 3 months Scr was 180 μmol/L, with no signs of hemolysis and the patient remained asymptomatic over one year of follow-up.
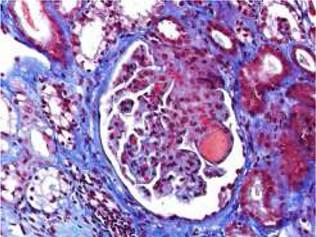
Fig. 1 Acute TMA showing thrombosis at glomerular capillaries and mesangiolysis (Masson's trichrome stain).
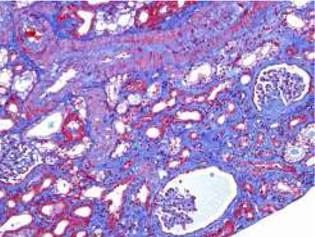
Fig. 2 Interstitial fibrosis of 5-10% and some microhemorrhages. Some ischemic and retracted glomeruli. Interlobar artery without acute thrombosis (Masson's trichrome stain).
In contrast to previous reports9,10 in our case the appearance of aHUS was late, at seven moths post-transplant, was not related with rejection nor with high plasmatic levels of tacrolimus. The patient did not show identified mutations and did not respond to correction of potential triggers. The pancreatic graft function remained always normal. After administration of limited doses of eculizumab, the renal function required three months to achieve SCr levels of 180 μmol/L and then remained stable, without hemolytic activity or symptoms. This highlights the importance of an early diagnosis and promptly treatment with eculizumab in order to improve renal and systemic results. In our case limited doses of eculizumab provided sustained improvement. Further evidence is needed to establish the optimal dosing in different clinical settings.

