












 (pdf)
(pdf)




 SciELO
SciELO  Google
Google  SciELO
SciELO  Google
Google 
 Permalink
PermalinkNutrición Hospitalaria
ISSN 1699-5198 ISSN 0212-1611
Conferencia Especial
Biología de la pared vascular y síndrome metabólico
A. Esteller Pérez
Catedrático de Fisiología y Director del Centro Tecnológico Multimedia. Universidad de Salamanca.
Introducción
En los países llamados del "primer mundo" ha surgido durante las últimas décadas una alarma creciente ante las consecuencias negativas que tiene la obesidad para la salud. Así se han asociado a esta disfunción de los hábitos alimentarios patologías tan graves como algunos tipos de cáncer, la diabetes tipo II, la hipertensión arterial (HTA) y las llamadas enfermedades cardiovasculares (ECVs). Estas patologías son en su conjunto responsables de más del 60% de los fallecimientos en esta zona del mundo y general, además, un coste económico enorme. En mi opinión, en la raíz de este nuevo panorama sanitario se encuentran tres fenómenos confluyentes: los cambios en los hábitos alimentarios generados por los avances tecnológicos (cadenas de frío, tecnología alimentaria, etc.); la tendencia al sedentarismo y al consumismo alentados por la oferta de cierto tipo de ocio y por la publicidad y el alto nivel económico, respectivamente), y la ansiedad crónica que genera la necesidad de satisfacer toda apetencia, siempre que esté arropada por tendencias de grupo. La figura muestra el porcentaje de muertes por causas diferentes en el mundo desarrollado (fig. 1). Parece evidente que nos encontramos ante un fenómeno nuevo y multietiológico que ha sido inducido o potenciado por el estilo de vida de los países desarrollados.

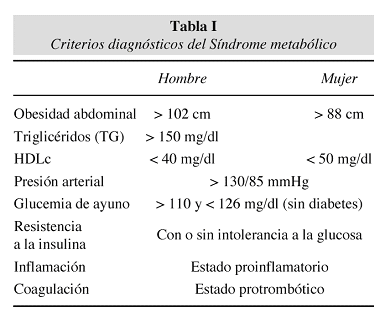
Se revisarán a continuación las bases fisiológicas y fisiopatológicas que podrían subyacer y explicar el fenómeno conocido como "síndrome metabólico" (hasta hace bien poco "Síndrome X"), que ha sido recientemente descrito de manera sistematizada (tabla I). Así se analizan, de forma necesariamente breve, las relaciones entre las células de la pared vascular y de la sangre, el desarrollo del proceso aterosclerótico y sus factores de riesgo más importantes, los principales mecanismos de daño endotelial, especialmente el estrés oxidativo y la disfunción endotelial consecuente, y por último las manifestaciones del síndrome metabólico y las bases genéticas desencadenantes del proceso.
Enfermedades cardiovasculares
El endotelio es, gracias a sus propiedades, un órgano determinante para el buen funcionamiento cardiovascular. Cuando su fisiología se altera por daño estructural o funcional, se inicia un largo proceso que puede desembocar en patologías tan graves como el infarto de miocardio, el ictus y la patología vascular periférica. De hecho, como ya se ha indicado, la principal causa de muerte, en las sociedades occidentales avanzadas, son las enfermedades cardiovasculares (ECV).
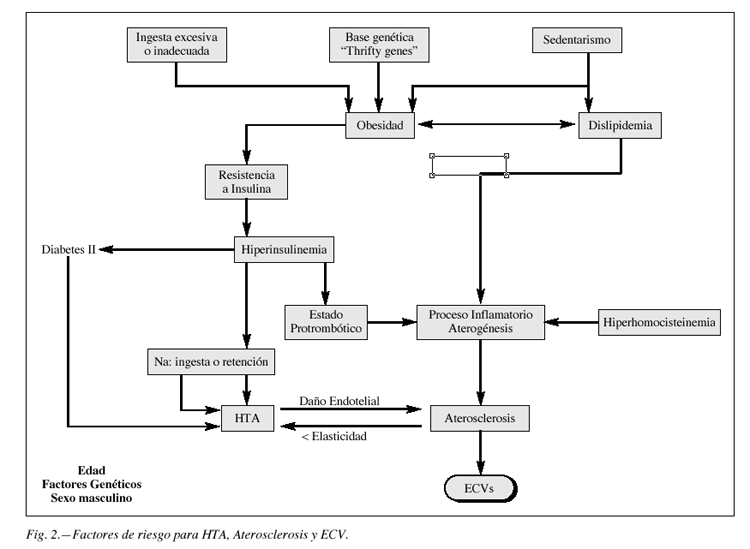
La expresión clínica de las enfermedades vasculares, aunque tengan un sustrato fisiopatológico común, depende del territorio en que se produzca la isquemia; la angina de pecho, el accidente cerebrovascular transitorio y la claudicación intermitente de las extremidades inferiores tienen una patogenia similar, pero son diferentes en sus manifestaciones. El factor patogénico más importante es la HTA, que potencia y conduce a la aterosclerosis, vía final común de toda la patología cardiovascular. Sobre estas situaciones actúan, con desigual intensidad, diversos factores de riesgo, congénitos o adquiridos (fig. 2).
El aumento de la incidencia de las ECV se ha frenado en la última década como consecuencia de los cambios en los hábitos de dieta y de actividad física. El mejor conocimiento de los mecanismos del estrés oxidativo y la forma de paliarlo han contribuido sin duda a esta evolución menos sombría.
Estructura vascular
Los vasos sanguíneos están formados por una capa adventicia y una capa media formada por células musculares lisas (CMLV) de potencia variable. Además, en la parte más interna se localiza la capa íntima formada por el endotelio de estructura variable según el tipo de vaso (arterial o venoso, grandes vasos, medianos o micovasculatura) y el territorio (cerrado, continuo, discontinuo o fenestrado).
Las células endoteliales forman una monocapa continua que tapiza la cara luminal interna de las arterias, las venas, los capilares y los vasos linfáticos, con una estructura muy organizada que asegura el acoplamiento funcional entre ellas. En el endotelio podemos encontrar dos zonas especializadas, la apical o luminal y la basal que interacciona con las proteínas de la matriz extracelular (MEC) de la lámina basal a la que está firmemente adherida, anclando las células al subendotelio. La MEC está compuesta fundamentalmente por clucoproteínas (laminina, fibronectina, vitronectina, trombospondina, entactina, heparan sulfato y factor Von Willebrand, entre otros).
Funciones del endotelio
El endotelio no expresa sus funciones de manera homogénea ya que existe una heterogeneidad que depende del tipo de vaso y del territorio en el que se encuentre. Así, por ejemplo, la permeabilidad es especialmente importante en los endotelios capilares y su intensidad está restringida por el tipo de endotelio. Así desde el endotelio cerrado de los capilares cerebrales, hasta el endotelio fenestrado del hígado encontramos una gración ascendente en la facilidad de paso de sustancias. Pero el endotelio vascular no es simplemente una barrera que separa la sangre de la pared vascular, sino también un importante órgano que está implicado en numerosas actividades por su capacidad de modificar su funcionalidad y regular la síntesis de diversos factores, en respuesta a cambios humorales, químicos o mecánicos en la sangre o en las células sanguíneas.
Así participa en diversas funciones de las que destacan, por su importancia para mantener la fisiología cardiovascular, las siguientes:
El mantenimiento del tono vascular y, por tanto, de la presión arterial, mediante la liberación de sustancias vasodilatadoras y vasoconstrictoras.
La capacidad de expresar moléculas de adhesión que a su vez controlan el reclutamiento de leucocitos al subendotelio, donde serán activados participando en el proceso inflamatorio.
La creación de una superficie no trombogénica por la presencia de cargas eléctricas negativas y por la síntesis de inhibidores de la agregación plaquetaria.
La síntesis y liberación de sustancias reguladoras del crecimiento del fenotipo de la migración de las células musculares lisas.
En estas funciones tienen un papel especialmente importante los factores sintetizados por el endotelio vascular (fig. 3).

Así en el tono vascular participan vasodilatadores como la PGI2, el NO o el factor hiperpolarizante derivado del endotelio (FHDE) especialmente importante en vasos pequeños, y también sustancias vasoconstrictoras como el tromboxano A2 (TXA2), la endotelina (ET1) o radicales libres de oxígeno (RLO). No hay que olvidar tampoco que una buena parte de la actividad de la enzima de conversión (ECA) y la producción de angiotensina II está asociada a las membranas plasmáticas de las células endoteliales.
En la actividad antiagregante y antitrobogénica participan el NO y la PGI2, así como moléculas como el heparán sulfato, la proteína C y el factor activador del plasminógeno (t-PA). Con actividad opuesta se pueden citar al TXA2, factor Von Willebrand (FvW), factor tisular (FT) y el inhibidor del activador de plasminógeno (PAI). La proliferación de las células de la pared, especialmente las CMLV, está especialmente regulada de manera inhibidora por el NO y la PGI2 y activadora por la endotelina 1 y la angiotensina II (fig. 4).

Por tanto, la fisiología vascular es dependiente de la integridad del endotelio porque mantiene un equilibrio entre la actividad biológica de sus factores y la de aquellos otros del mismo o distinto origen que la alteran. Entre las situaciones que más afectan a la estructura y actividad del endotelio, la aterosclerosis es, sin duda, la más importante.
Aterosclerosis
Denominamos aterosclerosis al engrosamiento y la acumulación focal de lípidos y macrófagos en la íntima arterial que origina un endurecimiento de la pared arterial. Se puede complicar con trombosis.
El término aterosclerosis no es sinónimo de arteriosclerosis, ya que éste engloba aquél y a:
La arteriosclerosis de Mönckberg se caracteriza por calcificaciones en la media de las arterias musculares. Al no producir estenosis, no hay clínica.
La arteriolosclerosis que se caracteriza por engrosamiento de la íntima (hialina o hiperplásica) de arterias pequeñas y arteriolas. Puede producir estenosis e isquemias localizadas (nefropatías).
La aterosclerosis es un proceso dinámico de la íntima arterial, durante el cual se establecen interacciones bidireccionales tanto con el endotelio y la sangre como con las células musculares lisas de la capa media. Las turbulencias del flujo sanguíneo en determinadas zonas de la circulación junto a fenómenos mecánicos debidos a la presión arterial condicionan la aparición de fenómenos adaptativos en la íntima, lo que explica que su grosor no sea uniforme y justifica la presencia de disfunciones locales en el revestimiento endotelial, que son la base del inicio del proceso de ateromatosis.
El inicio de las lesiones ateroscleróticas (fig. 5), xantoma de la íntima, suele producirse muy tempranamente, incluso durante la lactancia. Su distribución no coincide con los procesos ateroscleróticos posteriores lo que hace dudar que sea el auténtico origen de estas lesiones. El engrosamiento de la íntima se presenta como un trastorno de la función endotelial, sin cambios morfológicos manifiestos, caracterizado por una reactividad vascular alterada y una disminución de la síntesis de óxido nítrico. A ello se une el depósito de lipoproteínas procedentes de la sangre, engrosamiento patológico de la íntima, que se asocian a los proteoglicanos de la íntima y sufren procesos oxidativos y de glucosilación. La primera alteración macroscópica es la acumulación focal en la íntima de células espumosas, macrófagos y CMLV cargados de lípidos. Todo ello desencadena una respuesta inflamatoria, con una serie de procesos que van desde la expresión de proteínas quimiotácticas (MCP-1) y moléculas de adhesión (VCAM-1) a la producción de factores proliferantes (M-CSF) y de crecimiento y a la agregación plaquetaria, como se explica más tarde.

El ateroma tiene un núcleo lipídico rodeado de macrófagos, linfocitos T y miocitos. A partir del ateroma se forma el fibroateroma, en el que entre la luz vascular y el núcleo lipídico se produce una proliferación de miocitos sintetizadores de colágeno, de manera que la matriz extracelular de la íntima se ve sustituida por una fibrosis que engloba tanto las células como los lípidos del ateroma (síntesis de proteínas de la matriz extracelular). En esta fase se produce ya oclusión de la luz vascular, por lo que pueden aparecer manifestaciones clínicas isquémicas. Aparecen síntomas en el momento en que se desarrollan en la placa trombosis o hematomas que, al aumentar de forma aguda el tamaño de la lesión, producen una reducción brusca de la luz del vaso. En general, el proceso se inicia con la aparición de fisuras en la superficie luminal de la placa o incluso con su rotura, ya que se inhibe la síntesis y aumenta la degradación de las proteínas de la MEC (fig. 6). Este fenómeno puede dar lugar a la embolización de cristales de colesterol. En la evolución posterior aparecen fenómenos de cicatrización, calcificación o fibrosis, que estabilizan la lesión.

Disfunción endotelial
De forma general, se puede definir la disfunción endotelial como la serie de alteraciones que afectan la síntesis, liberación, difusión o degradación de los factores que se generan en el endotelio. Los mecanismos responsables de dichas alteraciones pueden originarse tanto por cambios en los receptores como de las señales intracelulares de transducción, o incluso por modificaciones en la respuesta de las células diana de dichos factores. La disfunción endotelial no es homogénea en sus características y su distribución, varía en función de la patología asociada, así como con el lecho vascular que se considere.
En la mayor parte de las lesiones ateroscleróticas, la función vascular del endotelio está atenuada, o incluso ha desaparecido. Las diversas formas de disfunción endotelial incluyen:
a) Menos liberación de NO, prostaciclina o EDHF.
b) Aumento de liberación de endoperóxidos.
c) Aumento de producción de radicales libres de oxígeno.
d) Aumento de liberación de endotelina.
e) Disminución de la sensibilidad del músculo liso vascular a los vasodilatadores de origen endotelial.
Tono vascular
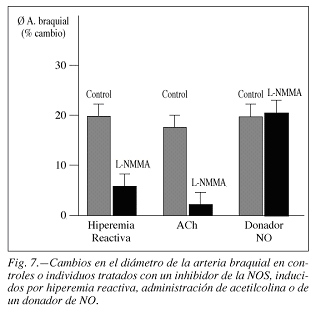
Diversos estudios han demostrado una menor respuesta vasodilatadora a la acetilcolina en aterosclerosis que en individuos controles, aunque la respuesta vasodilatadora al nitroprusiato sódico fue similar en ambos grupos. Estos datos sugieren que el mecanismo de relajación del músculo liso vascular dependiente de NO-GMPc no está alterado en la aterosclerosis, mientras que existe una alteración que afecta la producción, liberación o degradación del NO. Además, la respuesta constrictora al inhibidor de la síntesis de NO, L-NMMA, reduce el flujo sanguíneo braquial en mayor proporción en la aterosclerosis que en los controles.
La evaluación de la disfunción endotelial en relación con el tono vascular se puede realizar mediante técnicas de ultrasonidos (fig. 7) en arteria braquial.
Adhesión y activación de leucocitos
La disfunción endotelial también permite una mayor interacción de las plaquetas y monocitos con la pared vascular. La expresión de factores de reconocimiento y adhesión por parte de las células circulantes y de las endoteliales se activa tras daño endotelial o procesos inflamatorios. El tráfico de leucocitos en la microcirculación es crítica para la respuesta inmune normal de los tejidos. El reclutamiento de leucocitos está estrechamente regulado por la expresión secuencial de moléculas de adhesión específicas en la superficie de los leucocitos y de las células endoteliales. Las selectinas median la marginación y el rodamiento, mientras que las integrinas y las inmunoglobulinas permiten a los leucocitos la adhesión y la diapédesis.
La activación de los leucocitos modifica una serie de funciones vasculares, promoviendo la producción de eicosanoides, favoreciendo la adhesión celular y activando inicialmente la quimiotaxis. Además el leucocito activado participa en fenómenos de defensa activando la NOSi que permite producir de forma local cantidades importantes de NO. Este NO en exceso puede generar daño oxidativo que a su vez puede reducir la cantidad de NO de origen endotelial poniendo en marcha un círculo vicioso que hace procesar la lesión endotelial (fig. 8).

Agregación plaquetaria y proliferación celular
Cuando se produce una lesión vascular, además de la adhesión de las plaquetas en el lugar afectado, se pone en marcha el mecanismo de la coagulación tanto por la secreción tisular (FT) como por la exposición de la superficie desendotelizada. De esta forma, se activa la cascada enzimática de la coagulación que da lugar la aparición de trombina la cual actúa, por una parte, como potente activador plaquetario y, por otra, como catalizador de la formación y estabilización de redes de fibrina. La respuesta al estímulo de activación del sistema de la coagulación debe estar localizada, ampliada y modulada en el lugar del daño vascular.
Pero el daño endotelial, además del limitar la liberación de factores antiagregantes y antiproliferativos, favorece los procesos trombóticos de manera indirecta. Este fenómeno, junto con la activación de la proliferación de CMLV, hace avanzar el proceso aterogénico y acelera los procesos trombóticos (fig. 9).

Mecanismos de daño endotelial
El endotelio, como consecuencia de su localización anatómica, está expuesto a las fuerzas mecánicas sanguíneas. Esto es especialmente importante en las grandes arterias y especialmente en las zonas de bifurcación donde el flujo laminar se vuelve turbulento. Así la actividad mecánica de la presión arterial, responsable de un incremento de las fuerzas de cizalla sobre las células endoteliales vasculares, pasa a ser uno de los mecanismos inductores de la disfunción endotelial. Ello puede dar lugar a modificaciones estructurales y/o funcionales que afectarían a la producción o a la liberación de los distintos factores vasoactivos, así como a la respuesta de dichos agentes. La disfunción endotelial se manifiesta como una reducida respuesta vasodilatadora dependiente del endotelio, o una mayor respuesta constrictora dependiente o independiente del endotelio, como consecuencia de una alteración del equilibrio entre factores vasodilatadores y vasoconstrictores. Esto se asocia además a una elevación de las resistencias vasculares periféricas totales.
En resumen, la activación mecánica a corto plazo favorece la hiperpolarización de las CMLV y por tanto su relajación y activa la liberación de sustancias vasodilatadoras como NO y PGI2. A largo plazo favorece la producción de factores de crecimiento (PDGF), de factores procoagulantes (FT) y de moléculas de adhesión (Selectinas, integrinas, VCAM-1 e ICAM-1).
Daño oxidativo
El metabolismo oxidativo genera ATP en la cadena respiratoria de las mitocondrias, produciéndose agua como resultado de la adición de cuatro electrones al O2. Como subproducto se forma oxígeno molecular con electrones desapareados (fig. 10). En otras reacciones intracelulares se pueden formar peróxido de hidrógeno, radicales hidroxilo y aniones superóxido. Desde el mismo NO se pueden formar peroxinitritos y radicales hidroxilo. Las células son capaces de defenderse contra estos radicales deletéreos. Para ello utilizan reacciones, catalizadas por enzimas, que los transforman y eliminan. Además las defensas antioxidantes endógenas (glutation, bilirrubina, albúmina, ferritina, etc.), o exógenas (Vit. C, Vit. E, ß-caroteno, etc.), ayudan para evitar el daño oxidativo. No obstante, en determinadas situaciones de daño intenso o crónico, los radicales libres pueden causar peroxidaciones que dañan membranas, ADN, estructuras celulares y pueden producir la muerte celular.

Dislipemias y disfunción endotelial
La teoría que relacionaba la aterogénesis con niveles altos de lipoproteínas de baja densidad (LDL) ha sido sustituida por la que lo hace con la presencia de LDL oxidadas (LD-ox).
Hay muchas pruebas que relacionan la aterosclerosis con aumento de la peroxidación lipídica y del estrés oxidativo. Las lipoproteínas LDL, al ponerse en contacto con las paredes de las arterias, sufren una oxidación progresiva por parte de las células endoteliales, las CMLV y los macrófagos. La hipercolesterolemia aumenta tanto la cantidad de LDL que penetra en las paredes arteriales como su oxidación. La LDL oxidada (LDL-ox) es captada por los macrófagos, a través de receptores específicos de eliminación e induce la formación de células espumosas características de la aterosclerosis. Incluso las LDL mínimamente modificadas, no suficientemente oxidadas como para ser reconocidas por los receptores de eliminación, son capaces de afectar profundamente la expresión génica en células vasculares, produciendo una disminución en la producción de NO, aumentando el tono vascular y promoviendo el reclutamiento de monocitos y células T a través del aumento de expresión de moléculas de adhesión y factores quimiotácticos.
Las LDL-ox pueden, por múltiples mecanismos (fig. 11), modificar la estructura y la función de la pared vascular que conducen a la aparición de vasoconstricción, lesión endotelial, depósito lipídico, proliferación del músculo liso vascular y activación de la agregación plaquetaria. Las LDL-ox infiltran el espacio subendotelial donde se acumula y se capta por monocitos y macrófagos, que se transforman en células espumosas y posteriormente, en conjunción con otros factores, dan lugar a la formación de la estría grasa.
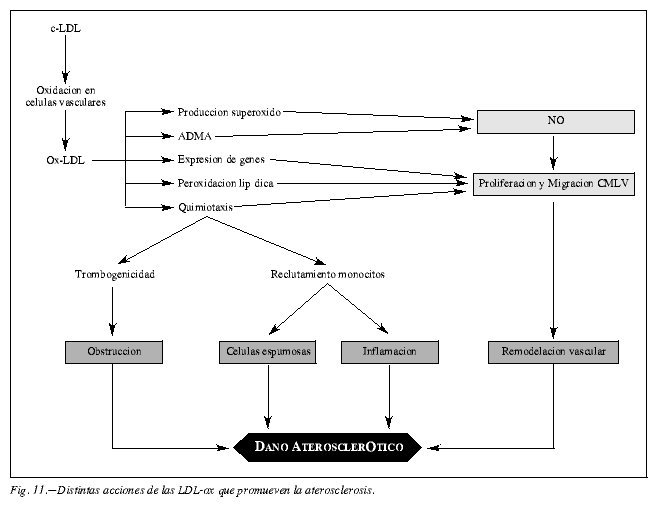
El mecanismo por el cual la producción de NO está reducido en hipercolesterolemia (LDL-ox) y en otras alteraciones metabólicas asociada con la aterogénesis parece ser multifactorial. Entre estos mecanismos se incluye el aumento de producción de radicales libres de oxígeno, la disminución de la síntesis de NO, y la acumulación de inhibidores endógenos del NO como la dimetil arginina asimétrica (DMAA). Esta molécula compite con la arginina reduciendo la síntesis de NO.
Asimismo, en eritrocitos y CMLV en cultivo, la presencia de LDL-ox modifica el transporte iónico transmembranario y aumenta la concentración de Ca++ libre citosólico, lo que probablemente explique el aumento de reactividad vascular en arterias.
Las LDL-ox también inducen apoptosis en las CMLV así como la transformación de un fenotipo contráctil a un fenotipo secretor, características de las células de la placa. Las LDL-ox son factores quimiotácticos para las CMLV y facilitarían su proliferación y posterior migración de la media a la íntima, contribuyendo no sólo al desarrollo de la placa arteriosclerótica, sino también al aumento de las resistencias vasculares periféricas y al remodelado vascular.
Los niveles elevados de LDL-ox, además de causar disfunción endotelial, alterar la función vascular y producir aterogénesis, favorece los procesos trombóticos al modificar el equilibrio entre factores protrombóticos y fibrinolíticos y al disminuir los niveles de NO induciendo el paso final del proceso aterosclerótico, la oclusión vascular (fig. 12).
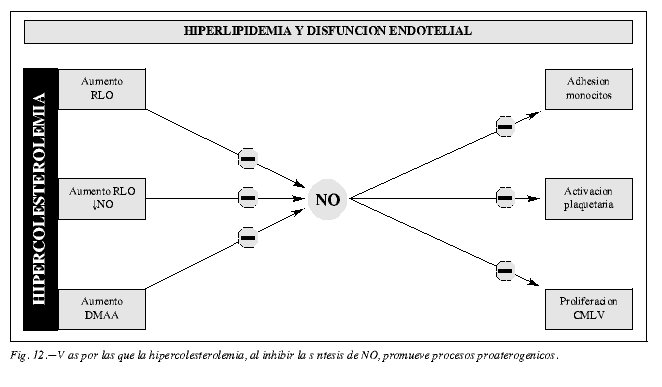
En conclusión, las LDL-ox contribuyen de manera significativa al aumento de la reactividad vascular, son potentes aceleradoras del proceso arteriosclerótico y facilitan los procesos de trombosis.
Hiperhomocisteinemia y disfunción endotelial
Desde hace muchos años se ha sugerido que la acumulación del aminoácido homocisteína es causa de daño endotelial y así se le ha señalado como factor de riesgo para el desarrollo de ECVs.
El metabolismo de la homocisteína es muy variado y en la mayor parte de las rutas existen enzimas que necesitan como cofactor vitaminas del grupo B (fig. 13).

Sus niveles en sangre se pueden alterar por muchas causas genéticas o adquiridas, modificables o no modificables (tabla II).

Se han descrito muchos mecanismos para explicar su papel en la disfunción endotelial y en la génesis de las ECVs (tabla III).

Papel sinérgico de la HTA y la hiperlipemia en la génesis de la disfunción endotelial
En la figura 14 se resumen las vías por las que la HTA y la hiperlipidemia se consideran dos factores de riesgo sinérgicos. Su presencia conjunta exige de una estructura terapéutica más potente para evitar la aparición de enfermedades cardiovasculares.

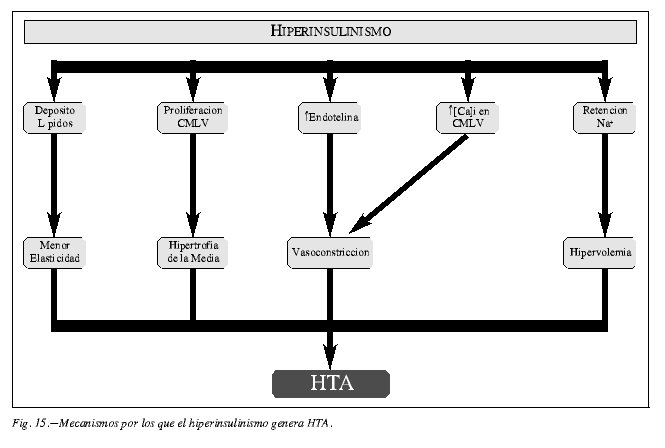
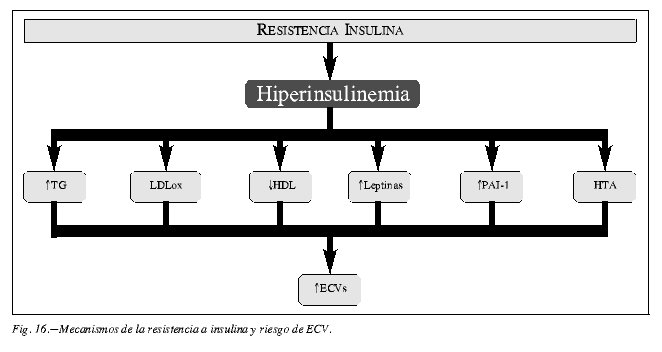
Obesidad, resistencia a la insulina e hiperinsulinismo en la génesis de la disfunción endotelial
En las figuras 15 y 16 se resumen las vía por las que estos tres factores de riesgo, encadenados temporalmente, promueven la disfunción endotelial y la incidencia de ECVs.
Receptores nucleares huérfanos (PPPARs) en la génesis de la disfunción endotelial
Los cambios en los hábitos de vida (sedentarismo e ingesta inadecuada o excesiva) y ciertos factores genético ("genes ahorrativos") se pueden considerar como factores desencadenantes de todos los procesos descritos. Se escapa a esta revisión su revisión. No obstante, y por su posible utilización terapéutica, sí se resume el papel de los receptores nucleares huérfanos denominados PPAR (tablas IV y V).


Resumen y recomendaciones
En este artículo se han revisado los factores de riesgo que participan en la génesis de las enfermedades cardiovasculares. Los fenómenos generados por los cambios en los hábitos de vida, junto con la influencia negativa de los denominados "genes ahorradores" que, en estas nuevas condiciones, son claramente desfavorables y explican la incidencia tan masiva de enfermedades cardiovasculares en el mundo occidental. En la etiopatogenia de las mismas destacan los fenómenos oxidativos de los lípidos de la pared vascular y las alteraciones dietarias que llevan al aumento de los niveles de homocisteína en sangre que, a su vez, potencian el primero alterando el equilibrio oxidantes/antioxidantes. Ante este panorama es imprescindible conseguir una modificación en los hábitos de vida que incluyan un control racional de los alimentos a ingerir, un incremento regulado de la actividad física y el abandono del consumo de sustancias tóxicas, entre las que el tabaco destaca por derecho propio.
Referencias
Díez J: "Enfermedad vascular e hipertensión arterial", Harcourt Brace, 1997.
Esteller A y Cordero M: "Fundamentos de Fisiopatología", 2.ª reimpresión. MacGraw Hill-Interamericana, 2002.
Goodnight SH y Hathaway WE: "Disorders of Hemostasis & Thrombosis", McGraw Hill. 2. Edition, 2001.
Greger R y Windhorst U: "Comprehensive Human Physiology: From Cellular Mechanisms to Integration", Springer-Verlag, 1996.
Levick JR: "An Introduction to Cardiovascular Physiology", 4.ª edición, Arnold Butterworths, 2003.
Loscalzo J, Creager MA y Dzau VJ: "Vascular Medicine: A Textbook of Vascular Biology an Diseases", Little Brown and Company, 1996.
McCully KS: "The Biomedical Significance of Homocysteine", Journal of Scientific Exploration 15, 5, 2001.
Sirica AE: "Cellular and Molecular Pathogenesis", Lippincott-Raven Publishers, 1996.
Tenenbaum A, Fisman EZ y Motro M: "Metabolic Syndrome and type 2 Diabetes mellitus: focus on peroxisome proliferator activated receptors (PPAR)", Cardiovascular Diabetology 2003, 2:4-11.
Correspondencia:
Alejandro Esteller Pérez
Centro Tecnológico Multimedia
Universidad de Salamanca Hospedería de Fonseca
C/ Fonseca, 2, 2.ª Planta
37008 Salamanca
Recibido: 25-V-2004.
Aceptado: 30-VI-2004.

