










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNutrición Hospitalaria
versión On-line ISSN 1699-5198versión impresa ISSN 0212-1611
Nutr. Hosp. vol.23 no.5 Madrid sep./oct. 2008
Síndrome de megavejiga-microcolon-hipoperistalsis intestinal: a propósito de un caso de supervivencia prolongada
The megacystis-microcolon-intestinal hypoperistalsis syndrome: a propos of a case with prolonged survival
M. I. Magaña Pintiado1, M. Al-Kassam Martínez1, C. Bousoño García2, E. Ramos Polo2 y M. E. Gómez Álvarez1
1Servicio de Farmacia. 2Servicio de Pediatría. Hospital Universitario Central de Asturias. Oviedo.
Dirección para correspondencia
RESUMEN
El síndrome de megavejiga-microcolon-hipoperistaltismo intestinal (MMIHS) es una grave enfermedad congénita autosómica recesiva, caracterizada por distensión vesical e hipoperistaltismo intestinal que provoca obstrucción intestinal funcional en el neonato, además de otras alteraciones. Presenta una incidencia muy baja, en torno al centenar de casos se describen en la bibliografía; la esperanza de vida apenas supera el año falleciendo generalmente por procesos sépticos. El caso que se presenta triplica esta supervivencia, con una calidad de vida y desarrollo ponderal muy aceptables. La nutrición parenteral domiciliaria unida a un seguimiento y coordinación multidisciplinar muy estrictos, constituyen las claves del éxito en esta patología.
Palabras clave: Síndrome de megavejiga-microcolon-hipoperistaltismo intestinal. Nutrición parenteral domiciliaria. Síndrome de Berdon. Colaboración multidisciplinar.
ABSTRACT
Megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS) is a severe congenital disease with autosomal recessive inheritance, characterized by vesical distension and intestinal hypoperistalsis what causes intestinal obstruction in newborn, with other abnormalities associated. It presents a low incidence, about a hundred cases are reported in the bibliography. Life expectancy doesn't reach a year because of the sepsis failure generally. In our study the survival is higher than the majority of the cases reported, with good cuality of life and acceptable ponderal development. Home parenteral nutrition with the following and multidisciplinary collaboration in a strict way, establish the success' key in this pathology.
Key words: Megacystis-microcolon-hypoperistalsis syndrome. Home parenteral nutrition. Multidisciplinary collaboration. Berdon syndrome.
Introducción
El síndrome de megavejiga-microcolon-hipoperistaltismo intestinal (MMIHS) o síndrome de Berdon, es una grave enfermedad autosómica recesiva con un pronóstico de vida inferior al año1,2. Es un hecho habitual que se detecte antes del nacimiento y se manifiesta rápidamente en el período neonatal inmediato, con una vejiga distendida y la disminución o ausencia de peristaltismo en el tubo digestivo. La obstrucción intestinal, habitual en estos pacientes, imposibilita la alimentación por vía enteral y provoca distensión abdominal y vómitos biliosos persistentes. Presenta una incidencia muy baja; el primer caso descrito por Berdon fue en 1976 y desde entonces, apenas se han publicado un centenar de casos. El pronóstico de esta enfermedad es grave y aunque existen algunos casos de supervivencia prolongada3-6, la mayoría de los afectados no superan el primer año de vida7,8, falleciendo habitualmente por complicaciones postoperatorias, secundarias a nutrición parenteral, sepsis e insuficiencia renal. En la estrategia terapéutica una de las claves es el soporte nutricional, sobre todo a través de la vía parenteral durante meses, lo que afecta en gran medida a la calidad de vida del paciente y de la familia.
En nuestro artículo, presentamos la evolución clínica y nutricional de un paciente pediátrico diagnosticado de Síndrome de Berdon, con una edad muy próxima a los 4 años y por tanto poco frecuente (fig. 1). Se describe el tratamiento con nutrición parenteral domiciliaria (NPD) que le permite un desarrollo adecuado, le aporta una calidad de vida muy aceptable y disminuye de forma considerable el coste asociado a una estancia hospitalaria prolongada. A través de la revisión de la historia clínica, se estudió la evolución de los datos antropométricos (peso y talla) así como los parámetros analíticos para su valoración: bioquímica completa, parámetros relacionados con el metabolismo del hierro y parámetros nutricionales específicos de la dieta (prealbúmina, transferrina, ácido fólico y vitamina B12). Adicionalmente, se evaluaron las complicaciones asociadas al soporte nutricional y la estrategia terapéutica en el contexto multidisciplinar.

Caso clínico
Varón de 42 meses, primer hijo de padres sanos no consanguíneos, diagnosticado en ecografía prenatal de ureterohidronefrosis bilateral y dilatación gástrica. En el período neonatal precoz únicamente llamó la atención, un abdomen globuloso y la primera deposición meconial al 4º día de vida, con ausencia absoluta posterior de deposiciones. Un enema opaco evidenció microcolon y malrotación intestinal, corrigiéndose éstaúltima quirúrgicamente. Una biopsia rectal fue positiva para enolasa neuronal y descartó aganglionismo. Se mantuvo en todo momento con nutrición parenteral, con intentos de ingesta oral que toleró finalmente a los 2 meses de vida con un volumen máximo de 7 ml/h. El paciente siempre presentó una diuresis adecuada, aunque mantenía las cifras de creatinina en límites altos de la normalidad. Posteriormente se le realizó pieloureterostomía bilateral mejorando su función renal. A los 20 días de vida presentó una sepsis nosocomial relacionada con el catéter por Staphylococcus epidermidis, tratada con teicoplanina y cefotaxima con buena respuesta9. Estos datos amplían lo ya publicado en el año 2004 por parte del Servicio de Neonatología y del Servicio de Cirugía Pediátrica del Hospital Universitario Central de Asturias10.
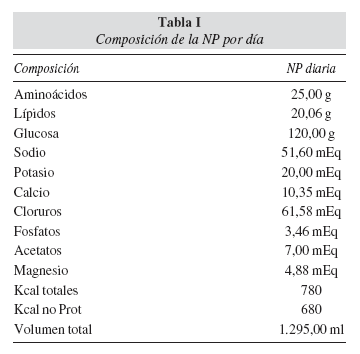
Actualmente el paciente presenta buen estado general, ureterostomía cutánea bilateral, peristaltismo y hábito intestinal conservados, relativamente bien nutrido y con un aceptable y progresivo desarrollo ponderal: percentil 10 de peso y talla, índice de Waterlow del 96%. Recibe nutrición parenteral (NP) desde el tercer día de vida y actualmente en régimen ambulatorio, suplementada con aportes orales a débito continuo, desde que inició tolerancia a los mismos. La composición de la NP por kg de peso y día es de 1,8 g de aminoácidos, 8,6 g de glucosa, 1,4 g lípidos, Na 3,7 mEq, k 1,4 mEq, Ca 0,7 mEq, Cl 4,4 mEq, P 0,2 mEq, Mg 0,3 mEq, contiene 55,7 kcal totales y un volumen total de 1.295 ml (tabla I). La NP es perfundida durante 17 horas, en pases de 40 ml/hora, con ascenso y descenso en una hora, siendo el tiempo de reposo sin NPD de 7 horas. Recibe tratamiento profiláctico frente al sobrecrecimiento bacteriano intestinal con metronidazol, además de trimetropim, fenobarbital, hierro, ácido ursodesoxicólico, ranitidina, eritromicina, vitamina E, vitamina D3 y darbepoetina. Mensualmente, se realiza control clínico, analítico (bioquímica completa, función renal, hemograma y reactantes de fase aguda) y control microbiológico de piel de ostomías y de catéter Hickman. Ha presentado episodios de obstrucción abdominal, uno de los cuales, secundario a bridas intestinales, requirió intervención quirúrgica. Las complicaciones relacionadas con la NP han sido dos: citolisis hepática transitoria, que remitió tras normalizar la relación de calorías procedentes de hidratos de carbono frente a grasas (60/40) (durante este período tenía niveles elevados de GOT, GPT, fosfatasa alcalina y bilirrubina, que se normalizaron) y una sepsis nosocomial relacionada con el catéter por Staphylococcus epidermidis, que respondió adecuadamente a tratamiento antibiótico.
Discusión
La etiología de este síndrome sigue siendo desconocida, se sospecha que presenta un patrón autosómico recesivo, dado que se han comunicado casos en hermanos gemelos y frecuente consanguineidad en las familias de los individuos afectados. Parece existir un predominio en el sexo femenino3,11 (4:1) y una clínica más grave en el sexo masculino2. En la mayoría de los casos publicados, la ecografía prenatal muestra un masa hipoecogénica intrapélvica, que puede deberse a quistes o masas abdominales, dilatación del estómago y la vejiga e hidronefrosis progresiva. Resulta complicado realizar con certeza un diagnóstico prenatal y se debe establecer el diagnóstico diferencial en primer lugar con causas de obstrucción mecánica congénita, obstrucción mecánica adquirida y obstrucción funcional y, en segundo lugar, con alteraciones funcionales (enfermedad de Hirschsprung, fármacos, alteraciones endocrinas, sepsis, etc.). La disfunción vesical que se manifiesta como megavejiga y la afectación intestinal, facilitan el diagnóstico frente al resto de patologías. A diferencia de la enfermedad de Hirschsprung, en la mayoría de los casos no se encuentran alteraciones ganglionares en los plexos mientéricos y submucosos. Clínicamente, como presenta el caso comunicado, es característica la distensión abdominal y el disminuido o ausente peristaltismo intestinal, acompañado en ocasiones de vómitos biliosos, incluso en ausencia de ingesta oral.
Lo complejo del síndrome provoca continuas y variadas complicaciones, por lo que la seguridad y la calidad de las terapias elegidas en cada circunstancia, tienen en común, una estrecha colaboración multidisciplinar y un soporte nutricional muy riguroso. El manejo de los catéteres y el cuidado de las vías son muy importantes ya que su uso será prolongado. Es imprescindible introducir precozmente la nutrición enteral, aunque sea de forma parcial, con el fin de evitar las consecuencias de una nutrición parenteral prolongada (riesgo de sepsis y fallo hepático); el tratamiento del sobrecrecimiento bacteriano con metronidazol y neomicina vía oral pueden disminuir el riesgo de sepsis4. Con el fin de evitar que las complicaciones derivadas de la NPD lleguen a poner en peligro la vida de estos pacientes, es fundamental un seguimiento clínico y nutricional por parte de todo el equipo multidisciplinar. La participación y coordinación de un equipo formado por: neonatólogos, pediatras, farmacéuticos, cirujanos, nefrólogos, genetistas, personal de enfermería, etc., y el adiestramiento de los cuidadores es imprescindible.
El trasplante multivisceral está indicado principalmente en aquellos pacientes con fallo hepático inducido por nutrición parenteral total4, lo que hasta ahora no ha sucedido en nuestro caso.
Entre los pocos casos de pacientes con supervivencia prolongada, como es el caso que se presenta, existe una dependencia absoluta a la nutrición parenteral total11, para conseguir que estos niños tengan calidad de vida aceptable, es imprescindible la hospitalización domiciliaria12. En nuestro caso, la nutricional domiciliaria permite el desarrollo del niño en su entorno familiar y social, y la posibilidad de poder plantearse la escolarización en un futuro no muy lejano, evitando además, los costes asociados a estancias hospitalarias prolongadas.
Referencias
1. Garber A, Shohat M, Sarti D. Megacystis-microcolon-intestinal hypoperistalsis syndrome in two male siblings. Prenat Diagn 1990; 10:377-87. [ Links ]
2. Annéren G, Meurling S, Olsen L. Megacystis-microcolonintestinal hypoperistalsis síndrome (MMIHS), an autosomal recessive disorder: clinical reports and review of the literature. Am J Med Genet 1991; 41:251-54. [ Links ]
3. Manop J, Chamnanvankij S, Wattanasarn C. Megacystis Microcolon Intestinal Hypoperistalsis Syndrome (MMIHS): A Case Report in Thailand. J Med Assoc Thai 2004; 87:1385-88. [ Links ]
4. Jiménez S, Moros M, Gimeno P, Castejón E, Ros L. Síndrome de megavejiga-microcolon-hipoperistaltismo intestinal: a propósito de un caso de supervivencia prolongada. An Pediatr (Barc) 2004; 60:369-72. [ Links ]
5. Srikanth MS, Ford EG, Isaac H, Mahour GH. Megacystis microcolon intestinal hypoperistalsis syndrome: late sequelae and possible pathogenesis. J Pediatr Surg 1993; 28:957-59. [ Links ]
6. Ciftci AO, Cook RC, Van Velzen D. Megacystis microcolon intestinal hypoperistalsis syndrome: evidence of a primary myocellular defect of contractile fiber synthesis. J Pedriatr Surg 1996; 31:1706-11. [ Links ]
7. Gillis DA, Grantmyre EB. Megacystis cicrocolon intestinal hypoperistalsis syndrome: survival of a male infant. J Pediatr Surg 1985; 20:279-81. [ Links ]
8. Masetti M, Rodríguez MM, Thompson JF, Pinna AD, Kato T, Romaguera RL y cols. Multivisceral transplantation for megacystis microcolon intestinal hypoperistalsis síndrome. Transplantion 1999; 68:228-32. [ Links ]
9. Suárez S, Calvo GR, Quiroga G, Díaz M, Moro B, López S: Síndrome de megavejiga-microcolon-hipoperistaltismo intestinal. Bol Pediatr 2004; 44:161-65. [ Links ]
10. Moreno V. Síndrome de megavejiga-microcolon-hipoperistaltismo intestinal. An Esp Pediatr 2001; 54:614-15. [ Links ]
11. Granata C, Puri P. Megacystis-microcolon-intestinal hypoperistalsis síndrome. J Pediatr Gastroenterol Nutr 1997; 25:12-19. [ Links ]
12. Rite GS, Fernández AS, Rebage MV, Marco TA, Esteban I, Romeo UM. Síndrome de megavejiga-microcolon-hipoperistaltismo intestinal. Ann Esp Pediatr 2000; 53:253-56. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
M. I. Magaña Pintiado.
Servicio de Farmacia.
Hospital Universitario Central de Asturias.
Oviedo.
E-mail: mmganyapintiado@yahoo.es
Recibido: 15-I-2008.
Aceptado: 12-V-2008.

