










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNutrición Hospitalaria
versión On-line ISSN 1699-5198versión impresa ISSN 0212-1611
Nutr. Hosp. vol.27 no.6 Madrid nov./dic. 2012
https://dx.doi.org/10.3305/nh.2012.27.6.6004
El factor de necrosis tumoral-α, la resistencia a la insulina, el metabolismo de lipoproteínas y la obesidad en humanos
Tumor necrosis factor-α, insulin resistance, the lipoprotein metabolism and obesity in humans
M.a M. Ramírez Alvarado1 y C. Sánchez Roitz2
1Departamento de Bioquímica. Escuela de Ciencias Biomédicas y Tecnológicas. Facultad de Ciencias de la Salud. Universidad de Carabobo. Valencia 2001. Estado Carabobo. Venezuela.
2Laboratorio Clínico César Sánchez Font. Centro Médico Dr. Rafael Guerra Méndez. Valencia 2001. Estado Carabobo. Venezuela.
Dirección para correspondencia
RESUMEN
En la obesidad el tejido adiposo produce moléculas proinflamatorias como el Factor de Necrosis tumoral-α, que tiene efectos locales en la fisiología del adipocito y efectos sistémicos en otros órganos. Muchos estudios relacionando TNF-α, obesidad, resistencia a la insulina y metabolismo lipídico se han realizado en ratas, conejos y perros, pero los resultados observados en varios de estos estudios han sido contradictorios y muchos de ellos no se han logrado reproducir en humanos, lo que hace difícil la interpretación del efecto del TNF-α sobre el metabolismo humano.
Objetivo: Realizar una revisión sistemática de los estudios realizados en humanos donde se relacione TNF-α, obesidad, resistencia a la insulina y metabolismo de las lipoproteínas.
Metodología: Se realizó una búsqueda en la base de datos PubMed de estudios realizados en humanos, tejidos humanos y líneas celulares humanas, relacionando TNF-α, obesidad, resistencia a la insulina y lipoproteínas.
Resultados: Existe una mayor producción de TNF-α en tejido adiposo de sujetos obesos. El TNF-α disminuye la respuesta celular a la insulina en adipocitos, hepatocitos y células musculares humanas. Hay un aumento de TNF-α en pacientes con dislipidemia, y la inactivación del TNF-α afecta el metabolismo lipídico. En hepatocitos humanos, el TNF-α inhibe la expresión de APO AI, lo cual puede disminuir la secreción de las lipoproteínas de alta densidad. El TNF-α afecta la excreción de colesterol al inhibir en hepatocitos a la enzima colesterol-7α-hidroxilasa.
Conclusión: El TNF-α disminuye la respuesta celular a la insulina, y tiene efectos sobre el metabolismo del colesterol y las lipoproteínas en humanos. Un mayor conocimiento de los mecanismos de la respuesta inflamatoria inducida por la obesidad en humanos, puede llevar a identificar nuevos blancos terapéuticos que permitan prevenir las complicaciones asociadas a la obesidad.
Palabras clave: Factor de necrosis tumoral-α. Resistencia a la insulina. Lipoproteínas. Colesterol.
ABSTRACT
In the obese adipose tissue produces proinflammatory molecules as tumor necrosis factor-α, which has local effects on adipocyte physiology and systemic effects in other organs. Many studies linking TNF-α, obesity, insulin resistance and lipid metabolism have been conducted in rats, rabbits and dogs, but the results observed in several of these studies have been conflicting and many of them have not been able to reproduce in humans, which on human makes difficult the interpretation of the effect of TNF-α on human metabolism.
Objective: To conduct a systematic review of human studies which relates, TNF-α insulin resistance and lipoprotein metabolism.
Methods: We searched the PubMed database for studies in humans, human tissue and human cell lines linking TNF-α, obesity, insulin resistance and lipoprotein.
Results: There is a increased production of TNF-α on adipose tissue of obese. TNF-α decreases the cellular response to insulin in adipocytes, hepatocytes and human muscle cells. There is an increase of TNF-α in patients with dyslipidemia, and inactivation of TNF-α affects lipid metabolism. In human hepatocytes, TNF-α inhibits expression of APO AI, which may decrease the secretion of high density lipoproteins. TNF-α affects the excretion of cholesterol by inhibiting the enzyme cholesterol-7α-hydroxylase in hepatocytes.
Conclusion: TNF-α decreases the cellular response to insulin, and has effects on the metabolism of cholesterol and lipoproteins in humans. A better understanding of the mechanisms of the inflammatory response induced obesity in humans, can lead to identifying new therapeutic targets that can prevent the complications associated with obesity.
Key words: Tumor necrosis factor-α. Insulin resistance. Lipoproteins. Cholesterol.
Introducción
La obesidad se define como un acúmulo de grasa corporal que se traduce en un aumento de peso. La obesidad es un factor de riesgo para enfermedades cardiovasculares, enfermedades pulmonares, enfermedades metabólicas, enfermedades osteoarticulares y para varios tipos de cáncer1-3. La obesidad, además de tener una fuerte asociación con la ocurrencia de condiciones médicas crónicas, tiene una asociación con el deterioro de la calidad de vida relacionada con la salud, y con un incremento en la atención sanitaria de los países.
La obesidad está relacionada con cambios en importantes parámetros fisiológicos como la presión arterial, sensibilidad a la insulina y la concentración sérica de lípidos. Se ha descrito una correlación positiva entre el grado de obesidad y ciertos desordenes asociados a la obesidad como la hipertensión arterial, dislipidemia e intolerancia a la glucosa2,4,5. Las causas de la obesidad y sus consecuencias a nivel fisiológico son objeto de estudio actualmente.
Recientemente, las dislipidemias, la resistencia a la insulina, la aterosclerosis y la obesidad han sido relacionadas con un estado de inflamación crónica. Las citoquinas no sólo son producidas por las células del sistema inmunológico, si no también por otros tipos celulares como lo adipocitos y las células no grasas del tejido adiposo. Además, estas citoquinas pueden actuar de manera local (autocrina/paracrina) y a nivel sistémico, y han sido implicadas en la disfunción del tejido adiposo y están relacionadas bioquímicamente con la resistencia a la insulina, la alteración en la liberación de ácidos grasos libres y el desarrollo de alteraciones del metabolismo hepático asociadas con la obesidad. Adicional a esto, el adipocito expresa receptores que le permiten responder a diferentes señales de regulación del metabolismo celular de las citoquinas6-8.
La demostración de que el tejido adiposo puede producir citoquinas tales como IL-6, IL-8, TNF-α y moléculas proinflamatorias como la proteína C reactiva (PCR), sugiere que los obesos pueden presentar un estado inflamatorio subclínico9-11. Existen evidencias que soportan la hipótesis que afirma que la obesidad es una condición inflamatoria que lleva a una activación crónica del sistema inmunológico innato lo cual conduce a las distintas condiciones clínicas que se observan en la obesidad. Estudios experimentales y la evidencia de estudios prospectivos y longitudinales en humanos son consistentes con el rol etiológico de la inflamación subclínica en la patogénesis de muchas de las enfermedades asociadas a la obesidad, como la aterosclerosis, donde la inflamación juega un papel clave en el inicio y progreso de la placa aterosclerótica12.
Muchos estudios relacionando TNF-α, obesidad y metabolismo se han realizado en ratas, conejos y perros, pero los resultados observados en varios de estos estudios han sido contradictorios y muchos de ellos no se han logrado reproducir en humanos. Por ejemplo, el tratamiento de la rata genéticamente obesa Zucker (fa/fa) con anticuerpos monoclonales neutralizantes anti-TNF-α revirtió el estado de resistencia a la insulina en estas ratas13, pero la administración de una sola dosis de anticuerpos anti-TNF-α neutralizantes a humanos obesos con diabetes tipo II14 o del receptor recombinante del TNF-α15 no produjo ningún efecto en la sensibilidad a la insulina. Este estudio tiene por objetivo hacer una revisión de los estudios que relacionen TNF-α, obesidad y metabolismo realizados en humanos, en tejidos humanos, así como en líneas celulares humanas.
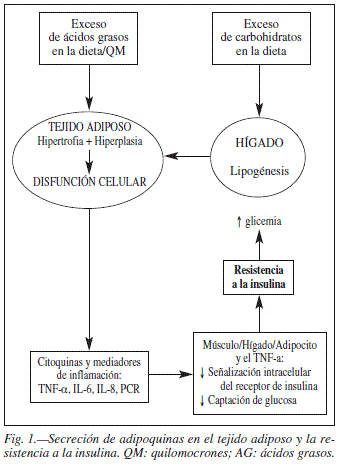
Hipertrofia e hiperplasia de los adipocitos
La principal función de los adipocitos es el almacenaje de los ácidos grasos en forma de triacilgliceroles (TAG). En sujetos obesos, el desbalance crónico entre las calorías consumidas y las gastadas causa un aumento de almacenamiento en forma de TAG en tejido adiposo, lo que se manifiesta de las siguientes formas: incremento de los lípidos intracelulares, aumento del tamaño de los adipocitos (hipertrofia), incremento del número de adipocitos (hiperplasia)16.
El incremento de lípidos dentro del adipocito, unido a la hipertrofia e hiperplasia del tejido adiposo, lleva a una disfunción celular que se manifiesta con anormalidades como la secreción de citoquinas y moléculas proinflamatorias por parte del tejido adiposo tales como TNF-α, IL-6, la proteína quimiotáctica de monocitos-1 (MCP-1), Factores Estimuladores de Colonia (CSF) y la sintetasa inducible de oxido nítrico (iNOS), generando el estado proinflamatorio descrito y reconocido en obesos. Sin embargo, estudios en los últimos años han puesto de manifiesto que los adipocitos no son la única fuente de secreción de citoquinas inflamatorias en el tejido adiposo. Las células no adiposas, que constituyen la fracción del estroma vascular, que incluye preadipocitos, células endoteliales, los fibroblastos, los leucocitos y macrófagos tienen parte de importante en la responsabilidad del desarrollo de la respuesta inflamatoria crónica observada en la obesidad17,18.
El TNF-α en la obesidad
El TNF-α es una citoquina proinflamatoria secretada en el sistema inmunitario por monocitos y macrófagos, por linfocitos T y B, células NK y por leucocitos polimorfonucleares. El TNF-α también puede ser secretado por otros tipos celulares como los adipocitos. El TNF-α tiene un amplio rango de efectos biológicos que incluyen inducción de apoptosis, citotoxicidad de células tumorales, activación y diferenciación de monocitos, inducción de la diferenciación de precursores inmaduros a monocitos, aumento de la actividad parasiticida y bactericida de los macrófagos al inducir las vías del superóxido y del óxido nítrico, inducción de la expresión de moléculas de adhesión en células endoteliales favoreciendo la migración local de leucocitos, aumento del receptor de IL-2 en linfocitos T y por consiguiente aumento de la respuesta proliferativa a IL-2, aumento de la respuesta de los linfocitos B estimulados. Por otro lado, el TNF-α tiene efectos fisiopatológicos al ser secretado en grandes cantidades en enfermedades agudas y crónicas, sepsis, infecciones crónicas, inflamaciones crónicas y cáncer. El TNF-α es sintetizado como una proteína transmembrana no glicosilada de 26 kDa que contiene una secuencia hidrofóbica. Tras una proteólisis se produce un fragmento de 14 kDa y un fragmento de 17 kDa que es la forma circulante como homotrímero unido de forma no covalente. La enzima encargada de la proteólisis es una metaloproteasa llamada TACE (TNF-αlpha converting enzyme). El TNF-α tiene dos tipos de receptores, tipo 1 y tipo 2 que son expresados en muchas células, incluyendo los adipocitos19-21.
El primer estudio que relacionó la obesidad con el TNF-α fue publicado por Hotamisligil y cols.22, este estudio estableció el inicio de los estudios de TNF-α/inflamación en la obesidad. La expresión de las citoquina proinflamatoria TNF-α esta incrementada en adipocitos de sujetos obesos y de sujetos con resistencia a la insulina23. Además del incremento de la expresión del TNF-α en tejido adiposo de sujetos obesos, se ha descrito que los sujetos obesos presentan mayores niveles séricos de TNF-α24, y que la pérdida de peso en obesos reduce los niveles séricos de TNF-α25 y la expresión del mRNA del TNF-α en tejido adiposo26.
El TNF-α y la resistencia a la insulina
La insulina es una hormona liberada por las células beta pancreáticas en respuesta a niveles elevados de glucosa en sangre, controlando funciones energéticas críticas en el metabolismo de los glúcidos, de las proteínas y de los lípidos, además, promueve el crecimiento celular. La insulina actúa en el metabolismo lipídico promoviendo el anabolismo e inhibiendo el catabolismo: regula al alza la lipoproteína lipasa (LPL) y estimula la expresión intracelular de enzimas lipogénicas como Acetil-CoA carboxilasa y ácido graso sintetasa, además, la insulina inhibe la lipasa sensible a hormonas (LSH) inhibiendo de esta manera la lipólisis.
Las acciones de la insulina son mediadas por cascadas de señalización intracelular, en las cuales la fosforilación inicial del receptor en residuos de tirosina lleva a una serie de eventos de fosforilación/desfosforilación de cinasas de tirosina y serina/treonina. Estas cinasas son las responsables de transmitir la señal de la insulina para la regulación de eventos metabólicos dentro de la célula. La incapacidad de las células blanco de responder a la insulina, genera un estado conocido como resistencia a la insulina.
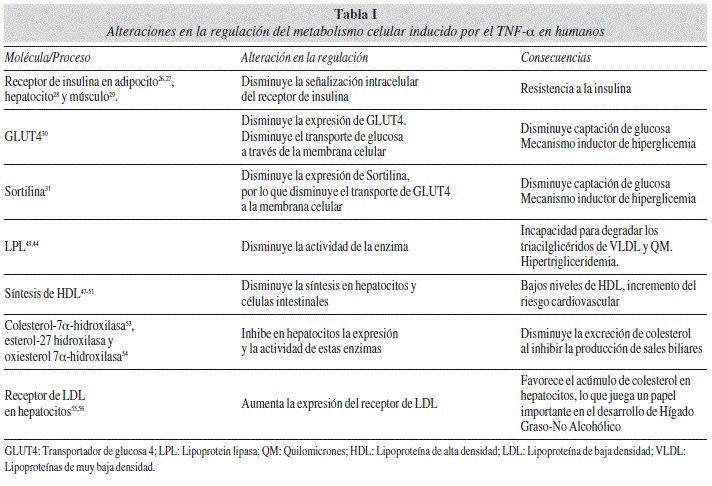
La expresión de TNF-α en los adipocitos se correlaciona positivamente con la obesidad y con la resistencia a la insulina. El TNF-α tiene múltiples efectos en el metabolismo, debido a un efecto paracrino sobre los adipocitos y a efectos en el metabolismo hepático. El TNF-α disminuye la señalización intracelular del receptor de insulina en adipocitos26,27, en células HepG2 (línea celular humana de carcinoma hepatocelular)28 y en células de músculo esquelético humano29, a través de la inhibición de IRS-1 (substrato del receptor de insulina-1) haciendo a esta molécula un pobre sustrato para fosforilación de los residuos de tirosina mediada por el receptor de insulina, y por tanto disminuyendo la amplificación intracelular de la señal del receptor insulínico, generando un estado de resistencia a la insulina, que trae consecuencias sobre el metabolismo celular.
El transporte de glucosa a través de la membrana celular se lleva a cabo por una familia de moléculas llamadas transportadores de glucosa (GLUT). El GLUT4 se encuentra almacenado en vesículas en el citosol de las células en el tejido muscular estriado, tejido muscular cardíaco y adipocito, siendo movilizadas las vesículas a la membrana celular por acción de la insulina, favoreciendo el movimiento de glucosa desde la sangre al interior de las células. Este comportamiento representa un mecanismo de regulación del metabolismo de la glucosa que sólo permite la entrada de glucosa al tejido muscular cuando es lo suficientemente elevada como para estimular la secreción de insulina y que en última instancia favorecerá la entrada del excedente de glucosa al interior celular. Se ha descrito que el tratamiento de adipocitos humanos en cultivo con TNF-α disminuye la expresión del mRNA de GLUT430. Los tejidos sensibles a la acción de la insulina, tales como el tejido adiposo, músculo esquelético y cardiaco, muestran incremento en la entrada de la glucosa a la célula cuando se estimula mediante insulina, debido a la activación del GLUT4. Esta activación está alterada debido a la exposición de las células al TNF-α, con disminución en la función de GLUT 4, el cual es un mecanismo inductor de hiperglicemia.
La sortilina es una proteína implicada en el trasporte de GLUT4 hacia la membrana citoplasmática. Los niveles de mRNA de la sortilina se encontraron disminuidos en tejido adiposo de pacientes diabéticos obesos, además, el tratamiento de adipocitos humanos en cultivo con TNF-α disminuye la expresión del mRNA de la sortilina, lo que se traduce en una menor translocación de GLUT4 hacia la membrana citoplasmática31. Estos resultados sugieren que la resistencia a la insulina en sujetos obesos se debe en parte, a la interferencia del TNF-α en expresión del gen y en la actividad de GLUT4 inducida por la insulina.
El TNF-α y su relación con el colesterol sérico total y las lipoproteínas
Existe una clara relación entre el TNF-α y los lípidos séricos, basados en estudios realizados en humanos. Primero, se ha descrito un aumento de TNF-α en pacientes con dislipidemia. En pacientes con hipercolesterolemia, se ha reportado elevados niveles séricos de TNF-α y acompañados de niveles elevados de triglicéridos (TG) y colesterol de lipoproteínas de baja densidad (LDL-C)32. Además, en pacientes hiperlipidémicos, los niveles séricos de TNF-α se correlacionan positivamente con los niveles de lipoproteínas de muy baja densidad (VLDL), TG y colesterol total (CT), y negativamente con el colesterol de lipoproteínas de alta densidad (HDL-C)33. En pacientes con hipercolesterolemia familiar hereditaria y con hiperlipidemia familiar hereditaria, afecciones que se caracterizan por niveles elevados de CT, LDL-C y un incremento significativo del riesgo de sufrir enfermedad coronaria prematura, se ha descrito un elevado nivel sérico de TNF-α34,35.
Segundo, la inactivación del TNF-α puede afectar el metabolismo lipídico. El boqueo de la unión del TNF-α a su receptor celular por medio de Ac. monoclonales o receptores solubles, es un tratamiento utilizado en enfermedades como la artritis reumatoide. El boqueo de la unión del TNF-α a su receptor celular provee evidencia directa del rol del TNF-α en el metabolismo lipídico. La administración de anticuerpos monoclonales anti-TNF-α humano (Infliximab) a pacientes con artritis reumatoide, aumenta los niveles de HDL-C y disminuye el índice aterogénico36,37, pero este es un efecto que sólo se mantiene en el corto plazo. Estudios más recientes, indican que la administración de Ac. anti-TNF-α monoclonales a largo plazo produjo un incremento de las concentraciones plasmáticas de CT y LDL-C y el índice aterogénico luego de 6 meses y 1 año desde el inicio de la terapia. Durante la terapia, los cambios en el estado inflamatorio se correlacionan negativamente con los cambios en los niveles de HDL-C y positivamente con la variación del índice aterogénico. Los autores concluyen que un año de tratamiento con Infliximab es probable que conduzca a un modelo más pro-aterogénico en las concentraciones delípidosplasmáticos38. En otro estudio similar, la administración de Ac. anti-TNF-α a pacientes con AR durante dos años produjo un incremento del CT, LDL-C y de los índices LDL/HDL y CT/HDL, y luego de 3 meses de tratamiento el HDL-C disminuyó significativamente39. Todo parece indicar que el TNF-α regula los lípidos séricos por medio de mecanismos complejos, por lo que se debe continuar estudiando el efecto del bloqueo del TNF-α sobre las concentraciones séricas de lípidos, con especial énfasis en los efectos a largo plazo de este bloqueo.
Tercero, la administración de estatinas a pacientes hipercolesterolémicos disminuyen los niveles séricos de TNF-α lo cual se ha descrito como un efecto no lipídico de las estatinas que juega un rol importante en la disminución del riesgo cardiovascular del paciente. Las estatinas son fármacos inhibidores de la enzima 3-hidroxi-3-metilglutarilcoenzima A reductasa (HMG-CoA reductasa), que bloquean la síntesis endógena de colesterol, lo cual disminuye los niveles séricos de colesterol. La simvastatina y la atorvastatina disminuyen los niveles séricos de IL-6 y de TNF-α en pacientes hiperlipémicos40. En otro estudio donde se determinó el efecto de la dieta y de la simvastatina sobre los marcadores serológicos de estrés oxidativo e inflamación, se obtuvo como resultado que comparado con la dieta sola, el tratamiento de los pacientes con dieta y simvastatina disminuyó significativamente los niveles de TNF-α de la molécula de adhesión intercelular tipo-1 (ICAM-1), de la PCR y del fibrinógeno41. También se ha reportado que el tratamiento con atorvastatina disminuye los niveles séricos de TNF-α, IL-1, IL-6, ICAM-1 y PCR en pacientes hipercolesterolémicos42.
Cuarto, el TNF-α inhibe la actividad de la LPL. La LPL es una enzima clave en la hidrólisis de los TAG de los quilomicrones (QM) y de las VLDL, ya que descompone los TAG a ácidos grasos libres y glicerol, liberándolos en músculo y tejido adiposo. En un estudio se determinó el efecto del TNF-α sobre la actividad de LPL en tejido adiposo humano en cultivo. La adición de TNF-α al medio de cultivo causó una inhibición dosis-dependiente de la actividad de la LPL, causada por una inhibición de los niveles de mRNA de la LPL y una inhibición en la síntesis proteica de esta enzima43. Este resultado ha sido confirmado por otros estudios44, e indica que el TNF-α es un potente inhibidor de la expresión del gen de LPL en tejido adiposo.
En estudios realizados en sujetos en un amplio rango de IMC, se determinó la relación entre la expresión del TNF-α y la actividad de LPL en el tejido adiposo. Los resultados indicaron que los sujetos obesos presentaron mayor expresión del TNF-α en comparación con las controles normopeso, y la expresión del TNF-α se correlacionó negativamente con la actividad de la LPL45,46. En muchos estudios se ha relacionado la obesidad con un estado de hipertrigliceridemia. La disminución de la actividad de la LPL puede explicar en parte la hipertrigliceridemia observada en obesos, debido a la incapacidad para degradar los triacilglicéridos de las VLDL y los QM.
Quinto, el TNF-α inhibe la expresión de APO AI y APO E en hepatocitos, interfiriendo en la síntesis de HDL. Las HDL son sintetizadas en hígado e intestino, siendo su función transportar el colesterol de los tejidos del organismo hasta el hígado para ser eliminado. Las APO AI y APO E son apolipoproteínas presentes en la estructura de las HDL. La APO AI participa activamente en el transporte reverso del colesterol y activa la lecitina-colesterol acil transferasa (LCAT), que cataliza la hidrólisis de un ácido graso de la fosfatidilcolina (lecitina) presente en las partículas de HDL en su estado naciente o discoidal y lo esterifica con el colesterol dando lugar al colesterol esterificado que pasa al interior de la partícula de HDL discoidal. El tratamiento con TNF-α de células humanas HepG2 en cultivo suprimió la expresión de la proteína APO AI y la secreción de APO E de una manera dosis-dependiente, además, también suprimió los niveles de mRNA de APO AI47,48. Estos resultados indican que el TNF-α puede estar implicado en la disminución de los niveles séricos de HDL al disminuir la síntesis de las apoproteínas que forman parte estructural de las HDL.
Sexto, el TNF-α disminuye la síntesis de HDL en células intestinales humanas. El tratamiento con TNF-α de las células Caco-2 en cultivo (línea celular de carcinoma de colon humano) inhibe la síntesis de HDL y de APO AI49, modulando el metabolismo lipídico intestinal.
Por otra parte, el transporte reverso del colesterol se ve facilitado por un transportador de la superficie celular llamada ABCA1 (ATP-binding cassette transporter A1). Este transportador media el paso limitante en la formación de HDL, promoviendo el flujo de colesterol y los fosfolípidos hacia la APO AI en las HDL49.Las mutaciones en ABCA1 son responsables del fenotipo de la enfermedad de Tangier. Este trastorno se caracteriza por una deficiencia severa de HDL, la acumulación de ésteres de colesterol en los macrófagos en diferentes tejidos, y aterosclerosis prematura50,51. Así, es evidente que ABCA1 es crítico para la síntesis de las partículas de HDL y desempeña un papel protector en la prevención de la aterosclerosis. Se ha descrito que el TNF-α disminuye la expresión de ABCA1 en las células Caco-2 en cultivo52, lo cual puede disminuir los niveles de HDL e interferir en el flujo de colesterol hacia las APO A1. La disfunción en la síntesis de HDL en hígado e intestino inducida por el TNF-α puede llevar a bajos niveles de HDL y a un incremento del riego cardiovascular, lo cual es de particular importancia en obesos.
Séptimo, el TNF-α puede afectar el metabolismo y le excreción de colesterol al inhibir en hepatocitos la expresión y la actividad de la enzima colesterol-7α-hidroxilasa, enzima clave de la síntesis de sales biliares a partir del colesterol en el higado53. Además, el TNF-disminuye la actividad de las enzimas esterol-27 hidroxilasa y oxiesterol 7α-hidroxilasa, las enzimas clave en la vía alternativa de síntesis de sales biliares54. El colesterol se elimina del organismo al ser convertido en ácidos y sales biliares las cuales son secretados en la bilis hacia el intestino para desecharse por heces fecales. Estos resultados indican que el TNF-α puede disminuir la excreción de colesterol al inhibir la producción de sales biliares, favoreciendo un acúmulo de colesterol que finalmente estará disponible para una mayor síntesis de lipoproteínas en el hígado.
Octavo, el TNF-α aumenta la expresión del receptor de LDL (LDL-r) en hepatocitos humanos, favoreciendo el acúmulo de colesterol en hepatocitos, lo que juega un papel importante en el desarrollo de la afección clínica Hígado Graso-No Alcohólico55,56.
Por lo hasta ahora descrito, se sugiere que el TNF-α tiene un profundo efecto sobre la respuesta celular a la insulina y sobre el metabolismo lipídico. Esto hecho es de particular importancia en los sujetos obesos, que presentan una mayor producción de TNF-α y un estado inflamatorio subclínico que juega un papel principal en el desarrollo de las enfermedades asociadas a la obesidad. Las investigaciones a realizar deben ir conducidas a determinar los distintos factores que inducen el estado de inflamación subclínica en sujetos obesos, lo cual puede llevar a nuevos blancos terapéuticos para prevenir las complicaciones relacionadas con los niveles elevados de TNF-α y con la obesidad.
Referencias
1. Caballero B. A nutrition paradox-underweight and obesity in developing countries. N Engl J Med 2005; 352: 1514-1516. [ Links ]
2. Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, Halsey J et al. Body-mass index and cause-specific mortality in 900000 adults: collaborative analyses of 57 prospective studies. Lancet 2009; 373: 1083-1096. [ Links ]
3. Samanic C, Chow WH, Gridley G, Jarvholm B, Fraumeni JF Jr. Relation of body mass index to cancer risk in 362,552 Swedish men. Cancer Causes Control 2006; 17: 901-909. [ Links ]
4. Huxley R, Mendis S, Zheleznyakov E, Reddy S, Chan J. Body mass index, waist circumference and waist: hip ratio as predictors of cardiovascular risk-a review of the literature. Eur J Clin Nutr 2010; 64: 16-22. [ Links ]
5. Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005; 111: 1448-1454. [ Links ]
6. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011; 121: 2111-2117. [ Links ]
7. Wellen EW, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 2003; 112: 1785-1788. [ Links ]
8. Yu YH, Ginsberg H. Adipocyte signaling and lipid homeostasis. Sequelae of insulin-resistant adipose tissue. Circ Res 2005; 96: 1042-1052. [ Links ]
9. Mohamed-Ali B, Gooddrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab 1997; 82: 4196-4200. [ Links ]
10. Bruun JM, Pedersen SB, Richelsen B. Regulation of interleukin 8 production and gene expression in human adipose tissue in vitro. J Clin Endocrinol Metab 2001; 86: 1267-1273. [ Links ]
11. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 1995;95:2409-2415. [ Links ]
12. Libby P. Inflammation in atherosclerosis. Nature 2002; 420: 868-874. [ Links ]
13. Hotamisligil GS , Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes 1994; 43: 1271-1278. [ Links ]
14. Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 1996; 45: 881-885. [ Links ]
15. Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 2000; 85: 1316-1319. [ Links ]
16. Salans LB, Cushman SW, Weismann RE. Studies of human adipose tissue. Adipose cell size and number in nonobese and obese patients. J Clin Invest 1973; 52: 929-941. [ Links ]
17. Cancello R, Clément K. Is obesity an inflammatory illness? Role of low-grade inflammation and macrophage infiltration in human white adipose tissue. BJOG 2006; 113: 1141-1147. [ Links ]
18. Bouloumié A, Curat CA, Sengenès C, Lolmède K, Miranville A, Busse R. Role of macrophage tissue infiltration in metabolic diseases. Curr Opin Clin Nutr Metab Care 2005; 8: 347-354. [ Links ]
19. Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol 1992; 10: 411-52. [ Links ]
20. Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annu Rev Cell Biol 1993; 9: 317-43. [ Links ]
21. Pennica D, Nedwin G, Hayflick JS, Seeburg PH, Derynck R, Palladino MA et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 1984; 312: 724-9. [ Links ]
22. Hotamisligil GS, Shargill NS, Spieeglman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259: 87-91. [ Links ]
23. Rotter V, Nagaev I, Smith U. Interlekin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulinresistant subjects. J Biol Chem 2003; 278: 45777-45784. [ Links ]
24. Olszanecka-Glinianowicz M, Zahorska-Markiewicz B, Janowska J, Zurakowski A. Serum concentrations of nitric oxide, tumor necrosis factor (TNF)-alpha and TNF soluble receptors in women with overweight and obesity. Metabolism 2004; 53: 1268-1273. [ Links ]
25. Dandona P, Weinstock R, Thusu K, Abdel-Rahman E, Aljada A, Wadden T. Tumor necrosis factor-alpha in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab 1998; 83:2907-2910. [ Links ]
26. Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H et al. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor expression. Gut 2010; 59: 1259-1264. [ Links ]
27. Fernández-Veledo S, Vila-Bedmar R, Nieto-Vázquez I, Lorenzo M. c-Jun N-terminal kinase 1/2 activation by tumor necrosis factor-alpha induces insulin resistance in human visceral but not subcutaneous adipocytes: reversal by liver X receptor agonists. J Clin Endocrinol Metab 2009; 94: 3583-3593. [ Links ]
28. Gupta D, Varma S, Khandelwal RL. Long-term effects of tumor necrosis factor-alpha treatment on insulin signaling pathway in HepG2 cells and HepG2 cells overexpressing constitutively active Akt/PKB. J Cell Biochem 2007; 100: 593-607. [ Links ]
29. Bouzakri K, Zierath JR. MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-alpha-induced insulin resistance. J Biol Chem 2007; 282: 7783-7789. [ Links ]
30. Hauner H, Petruschke T, Russ M, Röhrig K, Eckel J. Effects of tumour necrosis factor alpha (TNF alpha) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia 1995; 38: 764-771. [ Links ]
31. Kaddai V, Jager J, Gonzalez T, Najem-Lendom R, Bonnafous S, Tran A et al. Involvement of TNF-alpha in abnormal adipocyte and muscle sortilin expression in obese mice and humans. Diabetologia 2009; 52: 932-940. [ Links ]
32. Zubelewicz-SzkodziDska B, Szkodzi ski J, Danikiewicz A, Romanowski W, Bazelonis A, Muc-Wierzgon M, et al. Effects of simvastatin on pro-inflammatory cytokines in patients with hypercholesterolemia. Kardiol Pol 2003; 59: 465-474. [ Links ]
33. Jovinge S, Hamsten A, Tornvall P, Proudler A, Båvenholm P, Ericsson CG, et al. Evidence for a role of tumor necrosis factor alpha in disturbances of triglyceride and glucose metabolism predisposing to coronary heart disease. Metabolism 1998; 47:113-118. [ Links ]
34. Gokalp D, Tuzcu A, Bahceci M, Arikan S, Pirinccioglu AG, Bahceci S. Levels of proinflammatory cytokines and hs-CRP in patients with homozygous familial hypercholesterolaemia. Acta Cardiol 2009; 64: 603-609. [ Links ]
35. Pauciullo P, Gentile M, Marotta G, Baiano A, Ubaldi S, Jossa F et al. Tumor necrosis factor-alpha is a marker of familial combined hyperlipidemia, independently of metabolic syndrome. Metabolism 2008; 57: 563-568. [ Links ]
36. Popa C, Netea MG, Radstake T, Van der Meer JW, Stalenhoef AF, van Riel PL, et al. Influence of anti-tumour necrosis factor therapy on cardiovascular risk factors in patients with active rheumatoid arthritis. Ann Rheum Dis 2005; 64: 303-305. [ Links ]
37. Spanakis E, Sidiropoulos P, Papadakis J, Ganotakis E, Katsikas G, Karvounaris S, et al. Modest but sustained increase of serum high density lipoprotein cholesterol levels in patients with inflammatory arthritides treated with infliximab. J Rheumatol 2006; 33: 2440-2446. [ Links ]
38. Popa C, van den Hoogen FH, Radstake TR, Netea MG, Eijsbouts AE, den Heijer M, et al. Modulation of lipoprotein plasma concentrations during long-term anti-TNF therapy in patients with active rheumatoid arthritis. Ann Rheum Dis 2007; 66: 1503-1507. [ Links ]
39. Dahlqvist SR, Engstrand S, Berglin E, Johnson O. Conversion towards an atherogenic lipid profile in rheumatoid arthritis patients during long-term infliximab therapy. Scand J Rheumatol 2006; 35: 107-111. [ Links ]
40. Marketou ME, Zacharis EA, Nikitovic D, Ganotakis ES, Parthenakis FI, Maliaraki N, et al. Early effects of simvastatin versus atorvastatin on oxidative stress and proinflammatory cytokines in hyperlipidemic subjects. Angiology 2006; 57: 211-218. [ Links ]
41. Koh KK, Son JW, Ahn JY, Jin DK, Kim HS, Choi YM, et al. Vascular effects of diet and statin in hypercholesterolemic patients. Int J Cardiol 2004; 95: 185-191. [ Links ]
42. Ascer E, Bertolami MC, Venturinelli ML, Buccheri V, Souza J, Nicolau JC, et al. Atorvastatin reduces proinflammatory markers in hypercholestherolemic patients. Atherosclerosis 2004; 177: 161-166. [ Links ]
43. Fried SK, Zechner R. Cachectin/tumor necrosis factor decreases human adipose tissue lipoprotein lipase mRNA levels, synthesis, and activity. J Lipid Res 1989; 30: 1917-1923. [ Links ]
44. Hauner H, Petruschke T, Russ M, Röhrig K, Eckel J. Effects of tumour necrosis factor alpha (TNF alpha) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia 1995; 38: 764-771. [ Links ]
45. Bulló M, García-Lorda, Peinado-Onsurbe J, Hernández M, Del Castillo D, Argilés JM, et al. TNF-alpha expression of subcutaneous adipose tissue in obese and morbid obese females: relationship to adipocyte LPL activity and leptin synthesis. Int J Obes Relat Metab Disord 2002; 26: 652-658. [ Links ]
46. Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 1995; 95: 2111-2119. [ Links ]
47. Haas MJ, Horani M, Mreyoud A, Plummer B, Wong NC, Mooradian AD. Suppression of apolipoprotein AI gene expression in HepG2 cells by TNF alpha and IL-1beta. Biochim Biophys Acta 2003; 1623: 120-128. [ Links ]
48. Song H, Saito K, Fujigaki S, Noma A, Ishiguro H, Nagatsu T, et al. IL-1 beta and TNF-alpha suppress apolipoprotein (apo) E secretion and apo A-I expression in HepG2 cells. Cytokine 1998; 10: 275-280. [ Links ]
49. Mehran M, Seidman E, Marchand R, Gurbindo C, Levy E. Tumor necrosis factor-alpha inhibits lipid and lipoprotein transport by Caco-2 cells. Am J Physiol 1995; 269: G953-G960. [ Links ]
50. Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet 1999; 22:347-351. [ Links ]
51. Clee SM, Kastelein JJ, van Dam M, Marcil M, Roomp K, Zwarts KY, et al. Age and residual cholesterol efflux affect HDL cholesterol levels and coronary artery disease in ABCA1 heterozygotes. J Clin Invest 2000; 106: 1263-1270. [ Links ]
52. Field FJ, Watt K, Mathur SN. TNF-alpha decreases ABCA1 expression and attenuates HDL cholesterol efflux in the human intestinal cell line Caco-2. J Lipid Res 2010; 51: 1407-1415. [ Links ]
53. De Fabiani E, Mitro N, Anzulovich AC, Pinelli A, Galli G, Crestani M. The negative effects of bile acids and tumor necrosis factor-alpha on the transcription of cholesterol 7alpha-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4: a novel mechanism of feedback regulation of bile acid synthesis mediated by nuclear receptors. J Biol Chem 2001; 276: 30708-30716. [ Links ]
54. Memon RA, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. In vivo and in vitro regulation of sterol 27-hydroxylase in the liver during the acute phase response. potential role of hepatocyte nuclear factor-1. J Biol Chem 2001; 276: 30118-30126. [ Links ]
55. Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology 2008; 48: 770-781. [ Links ]
56. Liao W, Florén CH. Upregulation of low density lipoprotein receptor activity by tumor necrosis factor, a process independent of tumor necrosis factor-induced lipid synthesis and secretion. Lipids 1994; 29: 679-684. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
María Matilde Ramírez Alvarado.
Departamento de Bioquímica.
Escuela de Ciencias Biomédicas y Tecnológicas.
Facultad de Ciencias de la Salud. Universidad de Carabobo.
Av. Bolívar, Res. Santa Cecilia, PH-1.
Urb. El Recreo, Valencia 2001, Edo. Carabobo. Venezuela.
E-mail: mmramirez@uc.edu.ve / matilderamirez@hotmail.com
Recibido: 13-VI-2012.
Aceptado: 7-VIII-2012.

