










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNutrición Hospitalaria
versión On-line ISSN 1699-5198versión impresa ISSN 0212-1611
Nutr. Hosp. vol.28 no.3 Madrid may./jun. 2013
https://dx.doi.org/10.3305/nh.2013.28.3.6601
ARTÍCULO ESPECIAL
Progreso en el conocimiento de la microbiota intestinal humana
Progress in the knowledge of the intestinal human microbiota
Virginia Robles-Alonso y Francisco Guarner
Servicio de Aparato Digestivo. Hospital Universitario Vall d´Hebrón. Barcelona. España.
Dirección para correspondencia
RESUMEN
La aparición de nuevas técnicas de secuenciación así como el desarrollo de herramientas bioinformáticas han permitido no sólo describir la composición de la comunidad bacteriana que habita el tracto gastrointestinal, sino también las funciones metabólicas de las que proveen al huésped. La mayoría de los miembros de esta amplia comunidad bacteriana pertenecen a Dominio Bacteria, aunque encontramos también Archaea y formas eucariotas y virus. Únicamente entre 7 y 9 de las 55 Phyla del Dominio Bacteria conocidos están presentes en flora fecal humana. Su mayoría pertenecen además a las Divisiones Bacteroidetes and Firmicutes, encontrando también Proteobacteria, Actinobacteria, Fusobacteria y Verrucomicrobia. Bacteroides, Faecalibacterium y Bifidobacterium son los Géneros más abundantes aunque su abundancia relativa es muy variable entre individuos. El análisis metagenómico de la flora intestinal ha permitido describir una colección de 5 millones de genes microbianos que codifican para aproximadamente 20.000 funciones biológicas relacionadas con la vida de las bacterias. El ecosistema intestinal humano puede clasificarse en torno a tres grupos de acuerdo a la abundancia relativa de tres Géneros: Bacteroides (enterotipo 1), Prevotella (enterotipo 2) y Ruminococcus (enterotype 3). Estos grupos han sido denominados "enterotipos" y su descripción sugiere que las variaciones entre individuos están estratificadas. Una vez descrita la composición bacteriana sería interesante establecer la relación entre la alteración de equilibrios ecológicos con estados de enfermedad que puedan desembocar en una novedosa vía terapéutica.
Palabras clave: Enterotipo. Metagenómica. Microbioma. Simbiosis.
ABSTRACT
New sequencing technologies together with the development of bioinformatics allow a description of the full spectrum of the microbial communities that inhabit the human intestinal tract, as well as their functional contributions to host health. Most community members belong to the domain Bacteria, but Archaea, Eukaryotes (yeasts and protists), and Viruses are also present. Only 7 to 9 of the 55 known divisions or phyla of the domain Bacteria are detected in faecal or mucosal samples from the human gut. Most taxa belong to just two divisions: Bacteroidetes and Firmicutes, and the other divisions that have been consistently found are Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia. Bacteroides, Faecalibacterium and Bifidobacterium are the most abundant genera but their relative proportion is highly variable across individuals. Full metagenomic analysis has identified more than 5 million non-redundant microbial genes encoding up to 20,000 biological functions related with life in the intestinal habitat. The overall structure of predominant genera in the human gut can be assigned into three robust clusters, which are known as "enterotypes". Each of the three enterotypes is identifiable by the levels of one of three genera: Bacteroides (enterotype 1), Prevotella (enterotype 2) and Ruminococcus (enterotype 3). This suggests that microbiota variations across individuals are stratified, not continuous. Next steps include the identification of changes that may play a role in certain disease states. A better knowledge of the contributions of microbial symbionts to host health will help in the design of interventions to improve symbiosis and combat disease.
Key words: Enterotype. Metagenomics. Microbiome. Symbiosis.
Las nuevas tecnologías en el ámbito de la metagenómica
La aparición de las denominadas técnicas de secuenciación de alto rendimiento (high-throughput sequencing technologies) ha supuesto un punto de inflexión en la forma de entender la colonización bacteriana del intestino humano. Antes de la llegada de las citadas técnicas de secuenciación, el estudio de la diversidad bacteriana mediante el cultivo nos aportaba una visión sesgada de la composición bacteriana de la flora fecal, debido al desconocimiento de los requerimientos nutricionales de determinados subgrupos de bacterias y por ende la dificultad de cultivarlos en medios habituales.
La ventaja de las técnicas de secuenciación de alto rendimiento es su independencia del cultivo en medios biológicos, permitiendo una visión global a través del análisis del material genético presente en el medio que se quiera estudiar. Esta visión más amplia permite una descripción detallada de los diferentes miembros que forman la comunidad, bacteriana y de su abundancia relativa1.
Esta forma de abordaje ha llevado a acuñar el término metagenómica, definido como el estudio del material genético de las muestras recuperadas directamente de un determinado nicho ecológico2, y por tanto obviando la necesidad de aislamiento y cultivo individual de los distintos miembros. El metagenoma se describe como la colección de todo el material genético que constituye una comunidad ecológica. La aproximación más común consiste en la extracción del ADN de una muestra biológica, seguido de la amplificación y secuenciación de los genes que codifican para la subunidad 16S del ARN ribosomal. El gen 16S es común todas las bacterias y contiene regiones constantes y variables, por tanto, la similitudes y diferencias en la secuencia de nucleótidos del gen 16S permiten la caracterización taxonómica precisa de las bacterias que componen una comunidad, pudiendo discernir entre estratos de dominio y phylum hasta nivel de género y especie. La descripción del perfil taxonómico, se basa en la comparación de las secuencias del gen 16S de la muestra a estudiar con las secuencias de referencia de bases de datos.
Existe un abordaje todavía más integral, que consiste en la secuenciación génica de todo el ADN presente en una muestra. El abaratamiento del coste de las técnicas de secuenciación junto con el desarrollo de la genómica computacional ha hecho posible el análisis de mezclas complejas de ADN. De la información generada se puede inferir no solo información taxonómica, sino propiedades funcionales y metabólicas presentes en una comunidad bacteriana.
Durante los últimos años dos grandes proyectos a gran escala y dotados de elevados recursos económicos llevan cabo la tarea de descifrar la estructura y funcionalidad de la flora intestinal humana así como su relación con estados de enfermedad. Por una lado, el Proyecto MetaHIT financiado por la Unión Europea, y, en Segundo lugar, el Human Microbiome Project, subvencionados por el National Institute of Health de los Estados Unidos.
Diversidad y función de la microbiota gastrointestinal
Se estima que el colon alberga más de 1014 bacterias, en su mayoría pertenecientes al Dominio Bacteria. Aunque cabe destacar la presencia Archaeas metanógenas, eucariotas (levaduras y protistas) y virus en forma de fagos y virus animales.
Investigaciones basadas tanto en estudio del gen 16S como metagenómico sobre muestras fecales humanas han descrito representación de únicamente 7-9 de las 55 Phyla del Dominio Bacteria3-7. En concreto, más del 90% de las formas del Dominio Bacteria pertenecen a las Divisiones Bacteroidetes y Firmicutes. Otras Divisiones con abundancia destacable son Proteobacteria, Actinobacteria, Fusobacteria y Verrucomicrobia. Dentro del dominio Archaea encontramos reresentación de muy pocas especies, en su mayoría pertenecientes a Methanobrevibacter smithii.

Si estudiamos estratos taxonómicos más profundos, a nivel de especie, encontramos una gran riqueza a expensas de una gran variabilidad bacteriana individual, de forma que podemos considerar que cada individuo, es huésped de un perfil bacteriano único3. Además, el espectro de la comunidad bacteriana varía desde ciego hasta recto, de forma que podemos encontrar una diferente composición bacteriana dentro del mismo individuo, según analicemos una u otra región del colon. Sin embargo, cuando estudiamos la flora asociada a mucosa colónica, su composición parece mantenerse íntegra desde íleon hasta recto.
Se piensa que factores como la dieta, la ingesta de fármacos, viajes, o el mismo hábito deposicional, forman parte de las variables de determinan la ecuación que explica la composición de la flora fecal a lo largo del tiempo. Un reciente estudio8 recogió muestras de tres ubicaciones diferentes del organismo (intestino, boca y piel) de dos individuos sanos, a lo largo de un período de 15 y 6 meses respectivamente. Las conclusiones revelaron que la composición entre ubicaciones tiende a mantenerse estable a lo largo del tiempo, pero dentro de la misma localización corporal se detectó una baja estabilidad en la composición con respecto al tiempo. A nivel de Especie, muy pocos miembros bacterianos constituyen lo que ha venido a denominarse el núcleo del microbioma intestinal humano ("core human gut microbiota") ya que únicamente el 5% de las especies detectadas en muestras fecales, se mantenían presentes en todas las muestras tomadas a lo largo del tiempo en un mismo individuo8.
Analizando el gen 16S en muestras fecales en una cohorte de niños y adultos sanos procedentes de la zona amazónica de Venezuela, áreas rurales de Malawi y población urbana de Estados Unidos, se encontraron sorprendentes diferencias en la composición y diversidad de la colonización bacteriana entre los individuos de zonas con o sin desarrollo económico y social9. El análisis gráfico de composición sobre coordenadas reveló cómo las muestras procedentes de EEUU formaban grupos de asociación diferentes a las muestras procedentes de las otras dos regiones (Malawi y Amerindios). Se constató además en las tras poblaciones que la diversidad bacteriana se incrementaba con la edad, siendo las muestras procedentes de EEUU las menos diversas en comparación con las otras dos. Las diferencias entre poblaciones desarrolladas y no desarrolladas se relacionaban con diferentes factores de exposición ambiental (transmisión vertical y horizontal) así como con patrones dietéticos9.
El análisis metagenómico practicado sobre muestras fecales de una cohorte europea de adultos identificó un total de 3,3 millones de genes no redundantes4. Dicho estudio permitió además establecer el primer catálogo de genes microbianos procedentes del intestino humano. Se estimó que cada individuo albergaba una media de 600.000 genes en el tracto gastrointestinal, y 300.000 genes eran comunes al 50% de los individuos. De los genes identificados, el 98% eran bacterianos, y se describían entre 1.000 y 1.150 especies bacterianas, con una media por individuo de 160 especies6. Los Géneros más abundantes eran Bacteroides, Faecalibacterium, and Bifidobacterium si bien su abundancia relativa era muy variable entre individuos.

El Human Microbiome Project tenía por objeto analizar muestras de distintos ecosistema humanos: tracto gastrointestinal, piel, fosas nasales y tracto urogenital femenino en individuos sanos6,7. En el seno de dicho proyecto se describieron 5.177 perfiles taxonómicos microbianos así como más de 3.5 terabases de secuencias metagenómicas.
A nivel de tracto gastrointestinal El Human Microbiome Project amplió hasta 5 millones el catálogo de genes descrito previamente en el contexto del estudio MetaHIT. El cribado funcional en el seno de las técnicas de secuenciación de alto rendimiento consiste en la comparación de los genes derivados de la secuenciación íntegra de la muestra con secuencias que codifican para funciones conocidas. De esta forma es posible aproximarse a las funciones biológicas desempañadas por la comunidad bacteriana. Así se sabe que el amplio catálogo bacteriano descrito codifica proteínas implicadas hasta en 20.000 funciones biológicas4. Algunas de ellas, son necesarias para la autonomía bacteriana como las principales rutas metabólicas (metabolismo hidrocarbonado, síntesis de aminoácidos), o la propia expresión génica (ARN y ADN polimerasas, ATP sintasa). Otros genes codifican para funciones necesarias para la vida de las bacterias dentro del tracto gastrointestinal, es decir proteinas relacionadas con la adhesión a proteínas del huésped (colágeno, fibrinógeno, fibronectina) o el aprovechamiento de azúcares derivados de los glicopéptidos secretados por células epiteliales4.
Curiosamente, a pesar de la gran variabilidad interindividual en términos de taxonomía bacteriana el perfil genético funcional expresado por la comunidad bacteriana es bastante similar en individuos sanos7. Este concepto, parece clave a la hora de definir un ecosistema bacteriano sano, es decir, aquel ecosistema será tanto más "normal" cuanto más se parezca su perfil genético funcional a un estándar.
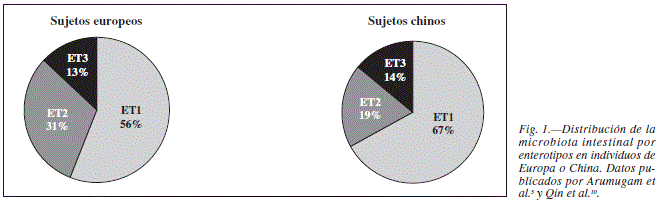
Enterotipos
Estudios recientes sobre diversidad bacteriana entre individuos sugieren que la flora intestinal humana puede agruparse de acuerdo a estados de equilibrio o de simbiosis, que han venido a definirse como "enterotipos"5. Cada uno de los grupos es diferenciable por la variación en cada uno de los 3 Géneros: Bacteroides (enterotipo tipo 1), Prevotella (enterotipo tipo 2) y Ruminococcus (enterotipo tipo 3). Esta categorización parece independiente de sexo, edad, nacionalidad o índice de masa corporal. Dichos hallazgos han sido descritos en el seno del proyecto MetaHIT y sobre población europea, americana y japonesa. La base para este agrupamiento es desconocida, sin embargo se especula con que pudiera estar relacionado con patrones dietéticos de larga evolución, ya que el enterotipo con predominancia del genero Bacteroides o tipo 1 ha sido asociado con dieta rica en proteínas y grasa, en contraposición al enterotipo tipo 2 (predominancia del genero Prevotella), más asociado al consumo de hidratos de carbono11. Estos resultados han sido reproducidos posteriormente sobre población china10. Otro reciente estudio basado en el análisis de muestras fecales en una cohorte de niños y adultos sanos procedentes de la zona amazónica de Venezuela, áreas rurales de Malawi y Estados Unidos9 encuentra el la misma agrupación por enterotipos en poblaciones originarias de áreas subdesarrollados, sin embargo, al incluir la población procedentes de Estados Unidos, esta clasificación pierde consistencia en adultos, apreciándose un intercambio entre los grupos Prevotella y Bacteroides. Por otro lado, parece que en población infantil la clasificación por enterotipos no tiene lugar5.
Disbiosis
Patologías como la enfermedad inflamatoria intestinal12, obesidad13,14, diabetes mellitus tipo 210, o colitis pseudomembranosa15 han sido asociados a cambios en la composición de la flora gastrointestinal, no obstante, la consistencia entre distintos estudios es aún pobre para algunas de ellas. El hecho de asociación no implica necesariamente causalidad, pudiendo ser estos hallazgos consecuencia de la propia enfermedad. Para establecer un papel etiológicos se precisan estudios de intervención y seguimiento con restauración de la diversidad o composición teóricamente perdidos. Es también necesario resaltar que cualquier aproximación terapéutica que intente devolver un equilibrio perdido ha de realizarse desde una óptica de ecología bacteriana, es decir, tratando de restaurar grupos bacterianos y no cepas aisladas16, tal como se ha demostrado en modelos animales17.
Este concepto se refuerza con el tratamiento eficaz de trasplante de flora fecal para el tratamiento de colitis pseudomembranosa refractaria18. Un estudio reciente llevado a cabo sobre individuos con síndrome metabólico y controlado contra placebo, procedió a la infusión de flora fecal procedente de individuos sanos delgados, y consiguió mejorar el perfil de resistencia insulínica a los pocos días de dicho trasplante19. Las implicaciones clínicas de estos cambios precisa de más estudios, aunque es claro que este abordaje emerge como una nueva vía terapéutica.
Conclusiones
La nuevas técnicas de secuenciación junto con el desarrollo de nuevas herramientas de análisis computacional permiten describir en profundidad la composición bacteriana del ecosistema intestinal humano así como conocer mejor las funciones que tal comunidad aporta al organismo anfitrión. Los siguientes pasos incluyen la identificación de los cambios que puedan estar asociados a determinados estados patológicos con el objeto de restaurarlos y restablecer la salud.
Referencias
1. Handelsman J, Rondon MR, Brady SF, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 1998; 5: R245-R249. [ Links ]
2. Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol 2008; 24: 4-10. [ Links ]
3. Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science 2005; 308: 1635-8. [ Links ]
4. Qin J, Li R, Raes J et al, MetaHIT Consortium, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464: 59-65. [ Links ]
5. Arumugam M, Raes J, Pelletier E, et al. MetaHIT Consortium: Enterotypes of the human gut microbiome. Nature 2011; 12: 473: 174-80. [ Links ]
6. Human Microbiome Project Consortium: Structure, function and diversity of the healthy human microbiome. Nature 2012; 486: 207-14. [ Links ]
7. Human Microbiome Project Consortium: A framework for human microbiome research. Nature 2012; 486: 215-21. [ Links ]
8. Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol 2011; 12: R50. [ Links ]
9. Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature 2012; 486: 222-7. [ Links ]
10. Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012; 490: 55-60. [ Links ]
11. Wu GD, Chen J, Hoffmann C, et al. Linking long term dietary patterns with gut microbial enterotypes. Science 2011; 334:105-8. [ Links ]
12. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 2012; 9: 599-608. [ Links ]
13. Ley RE, Turnbaugh PJ, Klein S, Gordon JI, et al. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444: 1022-3. [ Links ]
14. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature 2012; 489: 242-9. [ Links ]
15. Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet 2012; 13: 260-70. [ Links ]
16. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature 2012; 489: 220-30. [ Links ]
17. Manichanh C, Reeder J, Gibert P, Varela E, Llopis M, Antolin M, Guigo R, Knight R, Guarner F. Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res 2010; 20: 1411-9. [ Links ]
18. Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ: Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile -associated diarrhea. J Clin Gastroenterol 2010; 44: 354-60. [ Links ]
19. Vrieze A, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012; 143: 913-6. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Virginia Robles-Alonso.
Servicio de Aparato Digestivo.
Hospital Universitario Vall d´Hebrón.
Passeig Vall d´Hebrón, 119-129.
08035 Barcelona. España.
E-mail: vrobles@vhebron.net
Recibido: 28-III-2013.
Aceptado: 8-IV-2013.

