










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.18 no.8 ago. 2001
ORIGINAL
Enfermedad de Behçet: estudio retrospectivo
A. Baixauli, J. Calvo, J. J. Tamarit, C. Campos, S. García, A. Herrera
Sección Reumatología y Osteoporosis. Servicio Medicina Interna. Hospital General Universitario. Valencia
RESUMEN
Objetivo: Desde que se establecieron los criterios para el diagnóstico de la enfermedad de Behçet (EB), se han publicado numerosas manifestaciones clínicas asociadas a esta enfermedad. Intentamos estudiar la expresión de estas manifestaciones en 24 pacientes de nuestro hospital.
Material y métodos: Se diseñó un estudio retrospectivo desde 1990-2000 de los pacientes diagnosticados de EB en el Hospital General de Valencia. El diagnóstico de EB se realizó según los criterios de Grupo Internacional para el estudio de la enfermedad de Behçet. Los antecedentes personales, la expresión y evolución clínica fue revisada.
Resultados: De los 24 casos, catorce (58%) fueron varones y diez (42%) mujeres. La mediana de edad del diagnóstico fue de 33 años. Todos sufrían úlceras orales recidivantes. Las lesiones genitales aparecieron en 20 (83%) casos. Trece (54%) casos presentaron manifestaciones oculares, apareciendo las lesiones cutáneas en diez (42%) casos. Revisamos otras manifestaciones frecuentes en la EB: clínica articular apareció en 17 (70%) casos, clínica digestiva en seis (25%) casos, la clínica neurológica apareció en tres (12%) casos, la afectación vascular apareció en 2 casos (8%).
Conclusiones: Nuestros resultados son similares a los observados en otras series del área Mediterránea de la enfermedad de Behçet. Debe prestarse especial atención a las manifestaciones clínicas asociadas a la EB y que no están incluidas en los criterios diagnósticos.
PALABRAS CLAVE: Enfermedad de Behçet. Vasculitis.
Behçet´s disease: restrospective study
ABSTRACT
Background: Since the criteria for the diagnosis of Behçet's disease was established several isolated cases have been reported to the literature with clinical manifestations associated. We tried to analyze the clinical parameters associated to this disease in 24 patients in our hospital.
Methods: A retrospective analysis was made of the patients diagnosed of Behçet's disease at Hospital General of Valencia since 1990 to 2000. The diagnosis of Behçet's disease was made by the criteria of the International Study Group for Behçet disease. Data regarding personal, clinical manifestations and clinical course were collected.
Results: Of the 24cases, fourteen (58%) were men and ten (42%) were women. The median of the diagnosed was 33 years. All of them had oral recidivant ulcers. The genital ulcers appeared in twenty (83%) cases. Thirteen cases (54%) had ocular manifestations, and the skin lesions appear in 10 cases (42%). We review other clinical manifestations associated to Behçet disease: articular clinical appear in 17 cases (70%), digestive manifestations in six cases (25%), neurological manifestations three cases (12%), vascular clinical two cases (8%).
Conclusions: Our results are similar to the Mediterranean expression of Behçet's disease. We must study the clinical manifestations associated to this disease that are not include in the criteria of diagnosis.
KEY WORDS: Behçet's syndrome. Vasculitis.
Trabajo aceptado: 26 de Febrero de 2001
Correspondencia: A. Baixauli. Sección Reumatología y Osteoporosis. Servicio Medicina Interna. Hospital General Universitario. Avda. Tres Cruces, s/n. 46014 Valencia.
INTRODUCCIÓN
La enfermedad de Behçet (EB) agrupa un conjunto de manifestaciones que se caracteriza por la presencia de aftas orales, úlceras genitales múltiples, dolorosas y recurrentes, y por afectación ocular. Afecta principalmente a adultos jóvenes. Se considera una enfermedad autoinmune, ya que la principal lesión anatomopatológica es una vasculitis, que parece estar ligada a los aloantígenos HLA-B5 y DR5.
OBJETIVO
Exponer nuestra experiencia en la enfermedad de Behçet, comparar nuestra serie con otras revisiones de la literatura e incidir sobre los aspectos más importantes de esta enfermedad.
MATERIAL Y MÉTODOS
Estudio retrospectivo de los casos de enfermedad de Behçet atendidos en la sección de Reumatología del Servicio de Medicina Interna del Hospital General Universitario de Valencia. Se han revisado las historias de 24 pacientes diagnosticados de enfermedad de Behçet desde enero de 1990 hasta septiembre de 2000. El diagnóstico de enfermedad de Behçet se realizó siguiendo los criterios del Grupo Internacional para el Estudio de la Enfermedad de Behçet (1) (Tabla I).

RESULTADOS
En los resultados obtenidos se han encontrado un total de 24 enfermos, siendo 14 varones (58%) y 10 mujeres (42%), siendo la mediana de inicio de la enfermedad de 33 años, con un rango entre 18-59 años. El síntoma de debut de la enfermedad fueron las aftas orales en 21 ocasiones (88%), apareciendo las lesiones genitales en 2 (8%) ocasiones y el eritema nodoso en 1 ocasión (4%). Las úlceras orales recidivantes aparecieron en todos los casos (100%), mientras que las úlceras genitales recidivantes aparecieron en 20 ocasiones (83%). Se evidenció en 4 ocasiones orquiepididimitis. Las lesiones oculares aparecieron en 13 enfermos (54%), apareciendo en 3 ocasiones síntomas de sequedad ocular no filiada (23%), en 3 ocasiones uveítis anterior (23%), en 3 ocasiones papilitis (23%), en 3 ocasiones iritis (23%), tuvimos un (8%) paciente que presentó conjuntivitis. Las lesiones cutáneas aparecieron en 10 pacientes (42%), pseudofoliculitis se evidenció en 5 ocasiones (50%), eritema nodoso en 2 ocasiones (20%), acné corticodependiente en 1 ocasión, (10%). La prueba de patergia se realizó a 6 pacientes siendo positiva en una ocasión (16%). Las manifestaciones articulares aparecieron en 17 ocasiones (70%), evidenciándose sacroileitis en 7 pacientes (41%) y las artralgias en 4 ocasiones (23%), en 3 casos (45%) se evidenció sinovitis de rodilla, en 2 casos (12%) se cumplían, además criterios de espondilitis anquilosante un caso presentó osteítis condensans (7%). Las manifestaciones neurológicas se encontraron en 3 pacientes (12%), en forma de vasculitis cerebral. Las manifestaciones digestivas aparecieron en 6 pacientes (25%): 2 pacientes evidenciaron colelitiasis, aparecieron en 1 ocasión fisuras anales, ulceras ileales, duodenitis y colon irritable. A nivel vascular se recogen en dos pacientes tromboflebitis superficial con trombosis de vena cava superior y venas suprahepáticas en uno de ellos, presentando el otro trombosis del seno transverso, siendo en ambos casos la serología para el anticoagulante lúpico positiva. En un paciente se produjo la evolución hacia un mieloma (Tabla II).

DISCUSIÓN
La EB fue descrita inicialmente por H. Behçet, como un complejo trisintomático con úlceras orales y genitales e iritis recidivantes; posteriormente se le han añadido numerosas manifestaciones, fundamentalmente cutáneas, articulares, vasculares, neurológicas y gastrointestinales. (2-4)
Existe una descripción, que parece corresponder a la enfermedad de Behçet, hecha ya por Hipócrates en su tercer libro de epidemiología en el 5º siglo antes de Cristo: " Existen otros modos de fiebre... Muchos desarrollan úlceras aftosas. Ulceraciones en la zona genital... Oftalmia acuosa dolorosa de carácter crónico; excreciones fungoides en los párpados internos y externos que destruyen la visión de muchas personas" (5).
Existe un acuerdo fundamental en que el sustrato patogénico que motiva el polimorfismo típico de esta enfermedad es una vasculitis, donde predomina la afectación de vasos de pequeño calibre y que asienta preferentemente en capilares y vénulas, aunque también pueden afectarse venas y arterias. (6)
La distribución de la enfermedad es universal, aunque es mucho más frecuente en los países mediterráneos orientales y en Japón. En Japón se calcula una prevalencia de un caso por 10.000 habitantes (7). Turquía tiene la prevalencia más alta, entre 80-370 casos por 100.000 habitantes (5). En cuanto a la distribución por sexos, la enfermedad de Behçet es más frecuente en mujeres en Japón y Corea, siendo la edad de inicio más frecuente entre la tercera y la cuarta década de la vida (8).
Las series publicadas en la literatura española son escasas y con pocos enfermos. Eiroa et al, han calculado una prevalencia de 5,6 casos por 100.000 habitantes en el área sanitaria de La Coruña, con una incidencia anual de 0,53 casos por 100.000 habitantes para los varones y 0,32 casos por 100.000 habitantes para ambos sexos. La edad media en el momento del diagnóstico fué de 45.59 (+/-10,46) años. (9)
El Hospital General Universitario de Valencia abarca una población asistencial de 350.000 personas dentro del área 8 de la Comunidad Valenciana. La Sección de Reumatología tiene bajo su control a una población de 24 pacientes diagnosticados de enfermedad de Behçet según los criterios del Grupo Internacional para el Estudio de la Enfermedad de Behçet (en nuestro medio no se realiza de forma sistemática el test de patergia cutánea por lo que no será analizado). En nuestra revisión encontramos una mayor incidencia de la enfermedad en varones (59%) frente a las mujeres (41%) con una mediana de inicio de la enfermedad de 33 años. Estos parámetros se encuentran dentro de las características que presenta la enfermedad en la zona mediterránea. El Grupo Internacional para el Estudio de la Enfermedad de Behçet (GIEEB), propuso en 1990, unos nuevos criterios para el diagnóstico de la enfermedad (Tabla I). El diagnóstico diferencial de la enfermedad incluye la aftosis oral recurrente, infección por virus herpes simple, síndrome de Sweet, y enfermedades relacionadas con el antígeno HLA-B27, como puede ser la espondilitis anquilosante. En nuestro caso, aunque no ha sido objeto de este estudio hemos seguido durante años pacientes que presentan aftas orales de repetición y que no cumplen criterios de enfermedad de Behçet según el GIEEB. Estos casos han evolucionado hacia una aftosis oral recurrente o hacia un lupus eritematoso sistémico que debutó con lesiones orales (1,10).
Las aftas orales recidivantes, consideradas criterio diagnóstico indispensable por el GIEEB, suelen ser la manifestación inicial en el 70% de los casos. Las aftas orales son dolorosas, pequeñas (unos 10 mm de diámetro) con bordes bien definidos, y suelen durar 7-10 días curando sin dejar cicatriz y reapareciendo en intervalos variables. (3) En nuestra serie las aftas orales aparecieron en el 100% de los pacientes siendo el síntoma de debut de la enfermedad en el 88% de los casos. Al ser el criterio que permite el diagnóstico de la enfermedad, se encuentra en porcentajes cercanos al 100% en todas las series. Este signo puede preceder incluso años al resto de síntomas de la enfermedad. (11) El tratamiento con dosis bajas de corticoides (10-30 mg/ día de prednisona) suele suprimir las aftas orales y genitales. Para prevenirlas se pueden utilizar breves ciclos de prednisona oral, colchicina (0,5-1 g/ día) (12) o talidomida (100-300 mg/ día) (13).
Las úlceras genitales aunque pueden encontrarse en toda la zona perineal, suelen localizarse en el escroto y en el pene en los varones, mientras que en las mujeres se localizan en la vulva. Las características de las úlceras genitales son similares a las orales, aunque se describen de morfología más profunda y de bordes más irregulares, dejando una cicatriz residual tras su curación. En ocasiones pueden ser el signo de inicio de la enfermedad de Behçet, en nuestra revisión lo fue en el 8% de los casos, precediendo al resto de síntomas. Los sucesivos brotes de las úlceras genitales suelen asociarse a los brotes de las aftas orales. El tratamiento de las lesiones genitales es similar a las aftas orales (10,14).
La afectación ocular en la enfermedad de Behçet suele ser el síntoma de inicio en un 10% de los casos. Deben tenerse en cuenta como criterio diagnóstico cuando nos encontremos ante una uveítis anterior o posterior, presencia de células en el vítreo o bien vascularización retiniana. Los enfermos suelen presentar como síntomas más frecuentes la presencia de visión borrosa, dolor ocular, fotofobia, e hiperemia conjuntival. Puede existir afectación del segmento anterior (descripción clásica de la enfermedad) en forma de uveítis anterior e iridociclitis con hipopion o del segmento posterior en forma de uveítis posterior, coriorretinitis y vasculitis que puede dejar atrofia retiniana. La afectación ocular suele ser bilateral. En nuestra serie la afectación ocular se produce en 13 (54%) de los enfermos. Recogemos en nuestra revisión algunos síntomas que no se consideran criterios de enfermedad. La complicación más grave es la afectación de la retina que puede producir ceguera, no apareciendo en la evolución de ninguno de nuestros pacientes hasta la actualidad. Se calcula que cerca del 50% de los pacientes con afectación del segmento posterior están ciegos entre 4 y 8 años tras su evolución. El diagnóstico precoz de la afectación retiniana requiere la realización de la técnica de angiografía con fluoresceína. El tratamiento de la uveítis anterior se realiza con agentes midriáticos tópicos y colirios de corticosteroides. El tratamiento con esteroides orales no mejora el pronóstico visual y puede incrementar el riesgo de trombosis retiniana y cataratas. Se han utilizado inmunosupresores para intentar disminuir la dosis de esteroides como el clorambucil, la ciclosporina y la azatioprina. La ciclosporina A es útil hasta en el 80% de los enfermos con uveítis que no ha respondido a otras asociaciones, de todas formas su actividad y su nefrotoxicidad (ésta ultima poco frecuente en estos enfermos) es dosis dependiente. El tratamiento con interferón alfa ha presentado unas tasas de respuesta a nivel ocular del 95%, observándose remisión de los síntomas oculares en el 65% de los casos de los enfermos tratados con interferón alfa-2 a. (1,15-17).
En lo correspondiente a la afectación cutánea, el eritema nodoso es más frecuente en mujeres y suele localizarse en la cara anterior del tercio distal de las piernas; es indistinguible por su aspecto, etiología y evolución del eritema nodosa que se asocia a otras enfermedades. La pseudofoliculitis y los nódulos acneiformes son más característicos de la enfermedad de Behçet; siendo más frecuentes en varones y localizándose en la espalda, cara y cuello. Si el paciente es un adolescente o se encuentra en tratamiento con esteroides no se considera criterio diagnóstico de la enfermedad de Behçet (10,16).
El fenómeno de patergia, es un test de irritabilidad cutánea. Se realiza mediante la punción con una aguja estéril la cara anterior del antebrazo hasta 1 cm de profundidad. La lectura se realiza a las 24 y 48 horas. Es necesaria la aparición de induración y pústula para considerarlo positivo. No es exclusivo de la enfermedad de Behçet, pudiendo aparecer también en otras entidades como el síndrome de Sweet y el pioderma gangrenoso (14).
Las manifestaciones articulares, en forma de artralgias o artritis aparecen en cerca de la mitad de los pacientes. No forman parte de los criterios de la enfermedad de Behçet. Afecta a grandes articulaciones, sobretodo a rodillas, muñecas, tobillos y codos presentando un curso subagudo, intermitente y no deformante. La sinovial muestra el patrón de una artritis inflamatoria inespecífica. Las articulaciones sacroiliacas no suelen estar afectas por lo que no se la considera una espondiloartropatía. En nuestra serie recogemos un número sacroileitis asociada a la enfermedad de Behçet mayor que las registradas en las series clásica. La presencia de lesiones radiológicas en las exploraciones complementarias, radiología convencional y tomografía computarizada, permite diagnosticar en nuestro caso la sacroileitis en pacientes con mínima sintomatología. Las manifestaciones articulare suelen responder bien al tratamiento con aintiinflamatorios no esteroideos; el tratamiento con dosis bajas de esteroides, azatioprina, colchicina, talidomida e interferón alfa se han utilizado en el tratamiento de las artritis resistentes a antiinflamatorios no esteroideos (10,16,17).
La afectación vascular en nuestra revisión se asocia la presencia de anticoagulante lúpico con presencia de trombosis en grandes vasos venosos: venas suprahepáticas con afectación de la vena cava superior y la trombosis del seno transverso izquierdo. Ninguno de los casos cumplen criterios de lupus en la actualidad, cumpliendo ambos criterios de enfermedad de Behçet según la GIEEB. Bosch Gil y cols. realizó una revisión en lo que denomina "Angiobehçet", donde cifra la afectación vascular de la enfermedad de Behçet en el 30% de los casos, siendo la afectación del sistema venoso hasta siete veces más frecuente que la arterial; al mismo tiempo refleja que los pacientes con tromboflebitis superficial muestran una mayor tendencia a desarrollar trombosis venosas profundas y oclusión de vena cava (19). Ambos fenómenos aparecieron en nuestro paciente.
La afectación del sistema venoso se expresa en la enfermedad de Behçet como tromboflebitis superficial, trombosis venosa profunda, o trombosis de vena cava (20,21). Esta afectación venosa hace que se la deba tener en cuenta como causa potencial de trombosis venosa cerebral (22,23) y como posible afectación ocular en forma de trombosis de la vena central de la retina.
La afectación venosa puede manifestarse como síndrome de Budd-Chiari (como en nuestro caso), donde el determinante de la supervivencia será la extensión de la trombosis hacia en la cava inferior más que la presencia de trombosis de las venas suprahepáticas (24).
La afectación arterial es menos frecuente y puede manifestarse en forma de pseudoaneurisma cuyo riesgo de evolución hacia un aneurisma parece estar aumentado en esta enfermedad. La complicación de los pseudoaneurismas con trombosis es un hecho descrito en la literatura a pesar de no ser un hecho muy frecuente. La complicación trombótica arterial de forma aguda sin sospecharse lesión previa a nivel de la luz arterial en forma de pseudoaneurisma o placa de arteriosclerosis es muy rara, sin embargo este es un hecho que se postula en la trombosis de la arteria mesentérica de la que están descritos tres casos (25-27).
El tratamiento de las trombosis en la enfermedad de Behçet varía según las revisiones, no existiendo ningún tratamiento de elección. Se incluyen diferentes fármacos, entre los que se encuentran prednisona, heparina, ciclofosfamida, ciclosporina e incluso tratamientos combinados.(28)
La afectación neurológica es más frecuente en varones, sobretodo en aquellos en los que la enfermedad se inició en edades tempranas. Junto con las manifestaciones son las que producen mayor mortalidad. Clásicamente se ha descrito meningitis o meningoencefalitis, déficits neurológicos o síntomas psiquiátricos que pueden incluir cambios en la personalidad. La presencia de meningitis o meningoencefalitis linfocitaria suele aparecer durante los primeros años de la enfermedad, suele tener buen pronóstico y responde bien al tratamiento con esteroides, se han utilizado otros fármacos, como el clorambucil, pero en series cortas. La demencia aparece hasta en el 30% de los casos de enfermedad de Behçet (16,29).
Los síntomas intestinales aparecen hasta en el 50% de los pacientes con EB, siendo el síntoma más frecuente el dolor abdominal, seguido de nauseas, vómitos diarrea acompañada o no de restos de sangre, melenas, y estreñimiento. El hallazgo que encontramos en la mayoría de los casos son úlceras que pueden aparecer a lo largo de todo el tracto digestivo.
Las úlceras gastrointestinales en al EB, se localizan preferentemente en el íleon (75%), aunque también aparecen en el ciego (42%), colon transverso (13%), colon ascendente (13%), colon descendente (7%), sigma (5%) y recto (3%), siendo estos datos de la literatura japonesa. Existen casos en la literatura que describen lesiones a nivel del esófago.
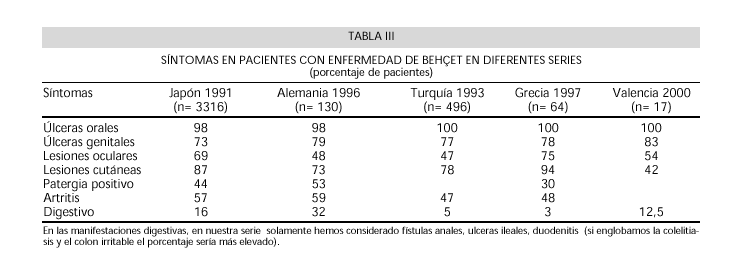
Nosotros encontramos manifestaciones digestivas en el 25% de los pacientes, sin embargo a la hora de relacionarlas con la enfermedad hemos excluido los diagnósticos de colon irritable y colelitiasis por considerar que era una asociación casual y que no estaba relacionada con la patogenia de la enfermedad. Así nuestra serie, tal como reflejamos en la tabla comparativa con las de otros países presenta clínica digestiva en un 12.5% de los casos, quedando limitada a afectación gastrointestinal (Tabla III).
Otras enfermedades que pueden presentar lesiones inflamatorias de las mismas características a las observadas en la EB son la enfermedad de Crohn, la colitis isquémica de predominio derecho, la tuberculosis intestinal y la infección por Yersinia spp., entre otras (30). En nuestra serie ninguna de estas enfermedades fue diagnosticada.
En la revisión realizada por J. Graña sobre la EB en Galicia su serie presenta un 40% de manifestaciones gastrointestinales, observándose en 5 casos úlceras recto cólicas, uno de ellos con fístula recto vesical.
Las úlceras ileocecales que se observan en la enfermedad de Behçet son largas y profundas y se sitúan en el área opuesta al mesenterio (rasgo característico de las lesiones intestinales que aparecen en la enfermedad de Behçet, aunque no exclusivo), perforándose con facilidad las de ésta localización; mientras que en la enfermedad de Crohn las úlceras son lineales, serpiginosas y con pseudopólipos. Existen pocos casos descritos en la literatura de evolución fatal de estas úlceras (31).
El diagnóstico diferencial suele estar claro en la mayoría de los casos, sin embargo hay pacientes con cuadros abigarrados que se sitúan entre la enfermedad inflamatoria intestinal crónica y la enfermedad de Behçet. La enfermedad inflamatoria intestinal y la enfermedad de Behçet comparten además gran número de manifestaciones extraintestinales como las úlceras orales, el eritema nodoso, la uveítis y la artritis. La histología muchas veces tampoco permite diferenciar ambas entidades. Suele utilizarse la presencia de granulomas en las úlceras intestinales como diagnóstico de enfermedad de Crohn y la presencia de una prueba de patergia cutánea positiva como signo de enfermedad de Behçet.
Zhiang aporta en su revisión una tabla que ofrecemos adaptada como ayuda para el diagnóstico diferencial de los síntomas y las úlceras en la enfermedad de Behçet (Tabla IV).

El tratamiento utilizado en la enfermedad inflamatoria intestinal también es útil en las lesiones gastrointestinales de la enfermedad de Behçet. La sulfasalazina y los corticosteroides son los fármacos más utilizados. Las dosis de corticoides está en función de la gravedad de la enfermedad. La complicación de estas úlceras, en forma de perforación o sangrado persistente, requiere tratamiento quirúrgico (32).
La afectación hepatopancreático o esplénica es poco frecuente. Se describen casos aislados de trombosis de las venas suprahepáticas en la enfermedad de Behçet (síndrome de Budd-Chiari). La presencia de una patrón analítico de colestasis disociada es frecuente en los períodos de actividad de la enfermedad (33). Se ha descrito algún caso de colestasis crónica y afectación intestinal donde se han podido objetivar alteraciones en los conductos biliares (34).
Así pues tras realizar una revisión la literatura parece ser que la enfermedad de Behçet engloba un conjunto de signos y síntomas, mayor que los que se expresaban en su descripción inicial, que son la expresión de una alteración en la inmunidad del enfermo que la sufre. Los tratamientos actuales han logrado mejorar el pronóstico de las formas más agresivas. Son necesarios estudios posteriores que nos ayuden a descifrar y entender la etiología y patogenia de la enfermedad para poder evitar las complicaciones asociadas a esta enfermedad.
Bibliografía
1. Criteria for diagnosis of Behçet's disease. International Study Group for Behçet disease. Lancet 1990; 335: 1078-1080. [ Links ]
2. Behçet H. Ubre rezidivierden aphtose durch ein virus versusch epaschwüre am mund, am auge und am den genitalien. Dermatol Woesmscher 1937; 105: 1152 1157. [ Links ]
3. Chajek T, Fairasu M. Behçet's disease: report of 4 cases and review of the literature. Medicine (Baltimore) 1975; 54: 179-196. [ Links ]
4. Smith GE, Kime LR, Pitcher JL: The colitis of Behçet's disease. A separate entity? Colonoscopia findings and literature review. Digestive Disease 1978; 18: 987-1000. [ Links ]
5. Kaklamani VG, Vaiopoulos G, Kaklamanis PG. Behcet's disease. Semin Arthritis Rheum 1998; 27: 197-217. [ Links ]
6. Bosch Gil JA, Clemente Rodríguez C, Sanchis Serrats J, de Sobregrau RC, Vilardell Torres M. Angiobehçet. Rev Clin Esp 1994; 194: 629-635. [ Links ]
7. Lehner T, Batchelor JR, Classification and immunogenetic basis of Behçet síndrome. En Lehner T, Barnes CG eds. Behçet's syndrome. Clinical and immunological features. New York: Academia Press 1970; 13-32. [ Links ]
8. Kastner DL. Intermitent and periodic arthritic síndromes. In: Koopman WJ, ed. Arthritis and allied conditions: a text book of rheumatology. 13th ed. Vol. 1. Baltimore. Williams & Wilkins, 1997: 1279-1306. [ Links ]
9. Eiroa P, Sánchez J, Rosales M et al. Estudio epidemiológico de la enfermedad de Behçet en el área sanitaria de La Coruña. Rev Esp Reumatol 1991; 18: 285-287. [ Links ]
10. Sakane T, Takeno, M, Suzuki N, Inaba G. Behçet's disease. N Engl J Med 1999; 21: 1284-1291. [ Links ]
11. Bang D, Hur W, Lee ES, Lee S. Prognosis and clinical relevance of recurrent oral ulceration in Behçet's disease. J Dermatol 1995; 22: 926-929. [ Links ]
12. Myyachi Y, Taniguchi S, Ozakki M, Horio T. Colchicine in the treatment of the cutaneous manifestations of Behçet's disease. Br J Dermatol 1981; 104: 67-69. [ Links ]
13. Hanza M. Treatment of the Behçet's disease with thalidomide (abstract 97). Royal society of Medicine international conference on Behçet's disease, September 5-6, 1985, London England. [ Links ]
14. Graña Gil J, Sánchez Meizoso MªO, Freiere González M, Galdo Fernández F. Enfermedad de Behçet. Seminarios de la Fundación Española de Reumatología. 1999; 1: 39-67. [ Links ]
15. Zouboulis CC, Orfanos CE. Treatment of Adamantiades- Behçet discase with systemic interferon alfa. Arch Dermatol 1998; 134: 1010-1016 [ Links ]
16. Graña Gil J, Galdo Fernández. F. Enfermedad de Behçet. En: Graña Gil J et al. Tratado Iberoamericano de Reumatología 1998; 520-529 [ Links ]
17. Gines MA, Jaber I, Pérez J, Estrada RV. Edema de papilla como primera manifestación de la Enfermedad de Behçet. An Med Interna (Madrid) 1996; 13: 255-256. [ Links ]
18. Benamour S, Zeroual B, Alaoui FZ .Joint manifestations in Behçet's disease: a review of 340 cases. Rev Rheum Engl ed 1998; 65: 299-307. [ Links ]
19. Fessler Bj. Thrombotic syndromes and autoimmune diseases. Rheuma Dis Clin North Am 1997; 23: 461-470. [ Links ]
20. Sagdiç K, Ozer ZG, Saba D, Türe M, Cengiz M. Venous lesions in Behçet's disease. Eur J Vasc Endovasc Surg 1996, 11: 437-440. [ Links ]
21. Campos C; Baixauli A; Calvo J; Glez.-Cruz MI; Herrera A. Síndrome vena cava superior en paciente con enfermedad de Behçet. Rheuma 2000, 4: 33-35 [ Links ]
22. Swedlow RH, Hanna Gr. Behçet's disease presentation with sagital sinus thrombosis diagnosed noninvasively. Headache 1996; 36: 115-118. [ Links ]
23. Farah S et al. Behçet's syndrome: a report of 41 patients witn emphasis on neurologycal manifestations. J Neurol Neurosurg Psychiatry, 1998, 64: 382-384. [ Links ]
24. Bayraktar Y, Balkanci F, Bayraktar M, Calguneri M. Budd-Chiari syndrome: a common complication of Behçet's disease. Am J Gastroenterol, 1997; 92: 858-862. [ Links ]
25. Akiyama K, Hirota J, Ohkado A,Shiina Y. Multivarious clinical manifestations of multiple pseudoaneurysms in Behçet's disease. J Cardiovasc Surg (Torino) 1998; 39: 175-178. [ Links ]
26. Yoshida S et al. Pseudoaneurysm of teh superficial femoral artery in Behçet's disease with spontaneous thrombosis followed by CT angiography. Cardiovasc Intervent Radiol 1998; 21: 342-344. [ Links ]
27. Mercié P et al. Thrombosis of the superior mesenteric artery and Behçet's syndrome. Rev Med Interne 1996; 17: 470-473. [ Links ]
28. Terzioglu E et al. Superior vena cava syndrome together with multiple venous thrombosis in Behçet's disease. Clin Rheumatol 1998; 17: 176-177. [ Links ]
29. Mateos-Colino A, González-Gay MA, Cerijo MJ, Santiago J, Rabuñal R, Brañas F. Neuro-Behçet: estudio evolutivo de cuatro casos tratados con clorambucil.An Med Interna (Madrid) 1995; 12: 600-602 [ Links ]
30. Okada M, Maeda K, Yao T, Iwashita A, Hoshiko K, Seo M Murayama H, Ohta K. Right- sided ulcerative colitis. J Gastroenterol 1996; 31: 717-722. [ Links ]
31. Martínez T, Colchero J, Conde J, Creagh R, Pujol. Complicación gastrointestinal grave de la enfermedad de Behçet. An Med Interna (Madrid) 1995; 12: 25-27. [ Links ]
32. Lee KS, Kim SJ, Lee BC, Yoon DS, Lee WJ, Chi HS. Surgical treatment of intestinal Behçet's disease. Yonsei Med J 1997; 38: 455-460. [ Links ]
33. Bayraktar Y, Ozaslan E, Van Thiel DH, Gastrointestinal manifestaions of Behçet's disease. J. Clin Gastroenterol 2000; 30: 144-154. [ Links ]
34. Hisaoka M, Haratake J, Nokamure. Small bite duct abnormalities and chronic intrahepatic cholestasis in Behçet's syndrome. Hepatogastroenterology 1994; 41: 267-270. [ Links ]

