










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.18 no.8 ago. 2001
REVISIÓN DE CONJUNTO
Hemoglobinuria paroxística nocturna.
Novedades patogénicas y terapéuticas
E. Sánchez Pérez, T. García Benayas, M. Bretón Arranz, C. Blasco Fanlo,
A. Poza Montoro, R. Blázquez Ruiz, S. Monzón Pérez
Servicio de Medicina Interna II. Hospital Clínico Universitario San Carlos. Madrid
RESUMEN
La hemoglobinuria paroxística nocturna (HPN) es la traducción clínica de una alteración de las células hematológicas, cuya etiopatogenia ha sido desconocida hasta hace relativamente poco tiempo. Intentamos resumir los aspectos más relevantes del proceso, y definir las características patogénicas, responsables de las manifestaciones clínicas de la enfermedad.
PALABRAS CLAVE: HPN. Etiología. Patogenia. Pig-A. Proteínas de anclaje del glucosilfosfatidilinositol.
Paroxysmal nocturnal haemoglobinuria. Pathogenic and therapeutic news
ABSTRACT
Paroxysmal Nocturnal Haemoglobinuria (PNH) is a clinical manifestation of an haemathology cell disease, whose etiology has been unknown for many years. We try to resume the most relevant facts of this entity and to define the pathogenesis which is responsible of the clinical manifestations of the disease.
KEY WORDS: HPN. Etiology. Pathogenesis. Pig-A. Glycosylphosphatidylinositol-anchored proteins.
Trabajo aceptado: 18 de Octubre de 2000
Correspondencia: Elena Sánchez Pérez. Paseo de los Melancólicos 22, 2º B. 28005 Madrid.
INTRODUCCIÓN
La HPN ha sido considerada una anemia hemolítica de causa intracorpuscular y predominio intravascular. Pero, hoy en día, se sabe que es algo mucho más complejo y se incluye dentro de los procesos denominados "panmielopatías clonales" (1). Se trata de un defecto adquirido de la célula madre pluripotencial de la médula ósea, que se transmitirá a las células que de ella desciendan, dando lugar al clon anormal característico de la entidad. Este clon puede surgir de una médula previamente dañada (lo que es más común, ya que es frecuente que se asocie a otros trastornos de células pluripotenciales) o de una médula normal (es decir, cariotipo normal) (2). Aparece principalmente en adultos jóvenes y afectando por igual a ambos sexos, con una incidencia aproximada de 1/100.000. Hasta hace pocos años, los conocimientos sobre la patogenia de la HPN eran muy limitados. Pero, durante ese tiempo, se han descrito los mecanismos de activación y regulación del complemento, las bases moleculares para explicar la exagerada sensibilidad al complemento de las células de la HPN y se ha identificado la mutación genética responsable. Todo este desarrollo ha permitido sentar la base patogénica del proceso.
ETIOLOGÍA Y PATOGENIA
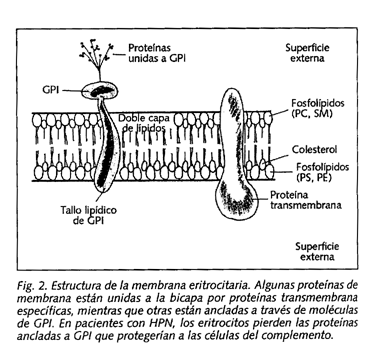
El trastorno básico de la HPN (responsable de la hemólisis intravascular característica) es la excesiva sensibilidad de las células hematológicas al complemento. Pero, ¿cuál es su base patogénica? Las células en la HPN carecen de sustancias "protectoras" capaces de inhibir la acción del complemento, por lo que incluso activaciones fisiológicas del mismo (tanto por vía clásica como por vía alternativa) producen la lisis de la membrana celular. Esas proteínas, encargadas de inactivar el complemento, se fijan a la membrana a través de una "molécula de anclaje", llamada glucosilfosfatidilinositol (GPI), que se inserta sobre la superficie celular. En la HPN no se produce la síntesis de GPI, por lo que éste no se "ancla" en las células hematológicas, dando lugar a una membrana anormal (Fig. 1).
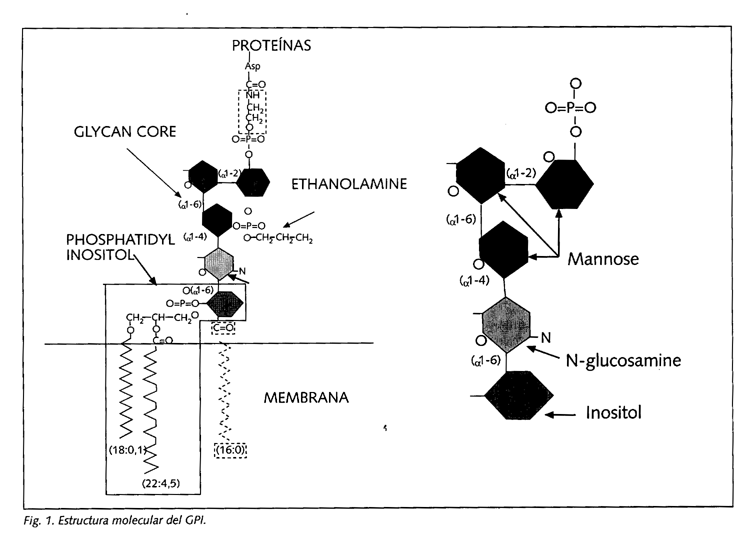
Todo ello es debido a una mutación adquirida en el gen encargado de codificar una enzima (parece ser una proteína de la familia de las glucosiltransferasas, pero aún no ha sido aislada), que es esencial en uno de los primeros pasos de la biosíntesis de la molécula GPI (3): es el gen glucosilfosfatidilinositol de la clase A (PIG-A=phosphatidylinositol glycan class A), localizado en el brazo corto del cromosoma X (Xp 22.1) en humanos (4).
La estructura molecular del GPI consiste en una molécula de fosfatidilinositol (PI), que es la parte que se inserta en la bicapa lipídica de la superficie celular, y un núcleo glicano (que consiste en una molécula de N-glucosamina, 3 moléculas de manosa y 1 de etanolamina). La manosa terminal es la que expone la amina, que reaccionará con el grupo carboxilo terminal de la proteína que se unirá a este receptor (Fig. 2).
El GPI es sintetizado en el retículo endoplásmico y productos de al menos 3 genes están involucrados. A través de estudios radiológicos del gen SPT 14 de las levaduras (que se considera que tiene un 45% de homología respecto al gen PIG-A humano), se ha comprobado que la N-acetilglucosamina no se incorpora al receptor. Esto parece indicar que el papel de la proteína PIG-A es transferir el azúcar al inositol.
En un intento de explicar totalmente la anormalidad de la cual resulta este síndrome, se han descrito las mutaciones somáticas producidas en la HPN y han surgido varios modelos, que pretenden dar una explicación:
1. La mayor parte son defectos debidos a la adición u omisión de 1, 2 o más nucleótidos. Este gen no codificaría ninguna proteína, lo que apoya el hecho de que la anormalidad más común de las células en la HPN sea la carencia total de GPI.
2. Las mutaciones identificadas (donde una base es sustituida por otra) producirán proteínas menos efectivas en la biosíntesis del receptor GPI, por lo que las células tendrán niveles intermediarios de proteínas ancladas al GPI.
3. Con algunas excepciones, las mutaciones son únicas.
4. En un número significativo de pacientes se han encontrado muchos clones (5). Se supone que es debido a mutaciones somáticas adicionales, dando lugar a subclones del clon original, lo que sugiere que estos sujetos tienen marcada tendencia a las mutaciones.
5. Se han encontrado mutaciones no significativas, que pueden funcionar para reducir, pero no abolir, la transcripción del gen PIG-A.
6. En los genotipos, sólo se ha encontrado un polimorfismo aislado (sustitución de arginina por triptófano en el locus 19).
7. En un solo caso, existían dos mutaciones en el mismo alelo.
8. Los defectos observados tanto en la HPN procedente de anemia aplásica, como en los que no proceden de ésta, son similares.
9. No ha habido diferencias entre los tipos de defectos encontrados en diferentes partes del mundo (hay más deleciones y adiciones de bases en la población tailandesa que en la japonesa, pero no es llamativo).
No está claro si el defecto en el gen PIG-A de las células hematológicas es condición suficiente para la manifestación del proceso. Pero, lo que sí está claro, es que ese defecto del gen PIG-A es el trastorno principal.
A partir de estos descubrimientos, se han iniciado un serie de investigaciones para determinar cuáles son las proteínas deficientes en estos pacientes (2). Las primeras alteraciones que se encontraron fueron la ausencia de dos enzimas:
--Fosfatasa alcalina leucocitaria: fue la primera anormalidad bioquímica descrita. Está también disminuida en las células de la leucemia mieloide crónica (LMC), pero algunos hechos marcan la diferencia:
1. En la HPN una proporción de neutrófilos parece no tener enzima y el resto parece tener cantidades normales. En la LMC todas las células tienen un grado variable de deficiencia.
2. La cantidad de RNAm que codifica la fosfatasa alcalina en los granulocitos de la HPN, está normalmente aumentada, mientras que en la LMC está marcadamente disminuida, indicando que la razón de la deficiencia es diferente en ambos trastornos.
--Acetilcolinesterasa eritrocítica: el grado de deficiencia de la enzima estaba directamente relacionado con el grado de sensibilidad al complemento, pero no se ha demostrado una relación causal.
Más tarde, se vio que otra proteína llamada FAD o CD55 (Decay Accelerating Factor), también estaba ausente. Fue la primera proteína que parecía tener un papel funcional en la patogenia del trastorno. Su función era disminuir la actividad del complejo C4b2a (una combinación de C4b y C2a que activa C3) con lo que se aceleraría la destrucción de C3. Varios estudios evidenciaron que la ausencia de FAD no era la única razón de la sensibilidad anormal de los hematíes al complemento. Es decir, debía haber un segundo defecto en las células de la HPN, que representara el papel fundamental en la patogenia de la enfermedad. Y éste es la proteína llamada CD59 (MIRL=Membrane Inhibitor of Reactive Lysis), cuya función era inhibir la formación del polímero C9 (complejo que produciría la disrupción de membrana). La deficiencia total o parcial de CD59 es la que explica la hemólisis característica del trastorno.
Posteriormente, se han descrito toda una serie de proteínas ausentes, alrededor de 20, aunque el número aumenta cada año:
--C8-Binding protein. Funciona de la misma forma que CD59. Algunos dudan sobre su existencia y de que sea distinta de CD59.
--CD58. (LFA-III). Proteína que interviene en las reacciones inmunes. Es un ligando para la adhesión de la molécula CD2 y la sujeción de las células T, que ocurre en la membrana linfocitaria. No está claro que esto traduzca una deficiencia inmunológica, posiblemente porque muchos de los linfocitos en la HPN no pierden proteínas unidas a GPI o porque la función puede llevarse a cabo por otros caminos.
--FCg-RIII (Fc receptor IIIa). Las células fagocíticas del sistema inmunológico tienen tres clases de proteínas, con capacidad para unirse a las porciones Fc de la molécula IgG, con lo cual se prepara el "blanco" para la fagocitosis. Las tres clases son : Fc RI, Fc RII, Fc RIII. En los neutrófilos, el mayor receptor de IgG es el Fc RIII unido a GPI, que falta en los neutrófilos de la HPN. El efecto de la deficiencia funcional de esta proteína está disminuido por la presencia de moléculas de Fc RII que son capaces de reemplazar la función de la proteína deficiente.
--UPAR (Receptor Urokinase Plasminogen Activator). Proteína presente, sobre todo, en los monocitos, cuya función es unir la enzima uroquinasa que activa el plasminógeno a plasmina en el inicio de la fibrinolisis. Esta función estará disminuida al faltar el receptor GPI (6).
--CD 14- Los polisacáridos de la pared celular bacteriana se unen a proteínas del suero, formando un complejo que, a su vez, se unirá al receptor CD14 de los monocitos y macrófagos y algunas células B. Cuando el complejo LPS-proteína se une a la célula, se produce la liberación de citocinas (particularmente TNF-a, IL-4, Il-6), que producirán la activación de las células endoteliales. C14 falta en los monocitos de la HPN, por lo que la respuesta de estas células a la endotoxina (LPS) está afectada. Se ha demostrado que en bajas concentraciones de LPS, los monocitos de la HPN muestran un marcado defecto en la producción de citocinas y además no regulan la expresión de otros CD, mientras que con concentraciones más altas los resultados son normales. Esto sugiere que CD14 es útil en la mediación de reacciones con LPS en bajas concentraciones, pero que otros caminos podrían mediar estas reacciones en concentraciones más altas.
--CDW52 (Campath-1): es una proteína portadora del antígeno Campath-1 presente en linfocitos, monocitos y neutrófilos. Los anticuerpos que reaccionan con este antígeno son capaces de activar el complemento, produciendo la lisis de la célula.. La molécula falta en las células de HPN, pero su efecto no está claro.
--CD24: se expresa, sobre todo, en las células B inmaduras. Su función no es conocida, al igual que el efecto de su deficiencia.
--CD48: antígeno leucocitario marcador de los linfocitos. Falta en las células de la HPN, pero la consecuencia de esto no se ha investigado.
--CD66: abarca un grupo de proteínas relacionadas con el antígeno carcinoembrionario (CEA). Su unión a ciertas proteínas produce la activación de los neutrófilos. Su efecto en la función celular se desconoce.
DEFICIENCIA DE PROTEÍNAS UNIDAS A GPI EN CÉLULAS SANGUENEAS DE LA HPN
Casi todas las proteínas ausentes en la HPN pueden ser detectadas con anticuerpos monoclonales, lo que ha permitido examinar la expresión de las proteínas sobre las diferentes líneas celulares (2):
PROGENITORES HEMATOPOYÉTICOS
Se distinguen por la presencia de marcadores asociados a la maduración. Las células "madre" de la médula normal llevan el antígeno CD34, pero no el CD38 (CD34+Cd38-), y además tienen CD55 y CD59. En la médula de la HPN se pueden encontrar células CD34+CD38- con y sin CD55 y CD59. La proporción de los precursores CD59 es paralela a la proporción de granulocitos defectuosos. Sin embargo, la proporción de células CD59+ parece ser mayor en los precursores hematopoyéticos de la sangre periférica que la encontrada en la médula. A veces, células deficientes en GPI, normalmente granulocitos, pueden ser detectados en la médula ósea varios meses antes de encontrarse en la sangre periférica. Esto ha sido llamado estadio pre-clínico de la HPN.
CÉLULAS DE LA SANGRE PERIFÉRICA
La proporción de células anómalas varía mucho de un paciente a otro, e incluso de un tipo de células a otro dentro del mismo paciente (2).
Eritrocitos
La proporción de hematíes con pérdida completa de proteínas unidas a GPI (Cél HPN III) suele ser discreta. La mayoría de los pacientes tienen menos del 80% de estas células. Las células con una deficiencia parcial (HPN II) están presentes aproximadamente en la mitad de los pacientes (en casi un 80%, representan menos del 20%).
La mayoría de los pacientes tienen un clon detectable de células con cantidades normales de proteínas unidas a GPI (Células HPN I), aunque el número puede ser muy pequeño.
Granulocitos
La mayoría de los pacientes tienen una proporción de granulocitos con pérdida completa de proteínas (cél HPN III) siempre mayor que en los hematíes. La proporción anómala probablemente refleja la proporción de células "madre" anormal. La vida media de los granulocitos anormales es normal, mientras que los hematíes anormales tienen una vida media acortada.
Linfocitos
El número de células B anómalas suele ser mayor que el de T, aunque esto no ocurre en todos los pacientes. A su vez, la proporción de células T anormales casi siempre es menor que la de granulocitos y está relacionada con la duración de la enfermedad. Las células anormales no responden a algunos estímulos. Pero, no está claro el efecto que la pérdida de proteínas unidas a GPI tiene sobre la función del sistema inmune, puesto que los pacientes con HPN no están totalmente inmunodeprimidos.
Monocitos
Su presencia se demuestra por la deficiencia del receptor CD14. La proporción anómala es similar a la de granulocitos. Los macrófagos no han sido examinados, pero como descienden de los monocitos, se presupone que también son deficientes.
Plaquetas
la pérdida de proteínas unidas a GPI tiene un gran efecto en su función. Las plaquetas parecen desempeñar un papel fundamental en la trombosis venosa que, sin duda, es la manifestación clínica más característica de la HPN y constituye una de las principales causas de muerte (7). Esto se explicaría por lo siguiente: las plaquetas de la HPN pierden CD55 y CD59, pero siguen siendo capaces de escindir los complejos de ataque del complemento en pedazos y formar vesículas, en cuya cara externa se encuentran los fosfolípidos a los que se fija la protrombina. Al no inhibirse la formación de complejos, brotan más vesículas, de lo que resultará un aumento de la actividad de protrombina y una gran tendencia a la agregación plaquetaria.
TRATAMIENTO
El tratamiento de la HPN ha sido empírico durante mucho tiempo, destinado a tratar las consecuencias más que las causas (8). Por tratarse de un trastorno de la célula pluripotencial (9), es lógico pensar que la única terapia curativa sería el trasplante de la médula ósea. En el momento actual, continúan las investigaciones sobre métodos para poder "separar" las células GPI+ del resto y realizar trasplantes autólogos (10).
Durante años, el pilar terapeútico de la HPN han sido los glucocorticoides (prednisona a dosis moderadas), que se sabe disminuyen la activación del complemento, con lo que se interrumpe la hemólisis. Para todos aquellos pacientes refractarios al tratamiento con glucocorticoides, se proponen como alternativa los andrógenos (concretamente el danazol), que aumentan la concentración de hemoglobina (12). Debido a sus efectos secundarios, este tratamiento se reserva para aquellos casos en los que las necesidades de transfusión son muy altas. Respecto a la anticoagulación, lo que está claro es que una vez que se ha producido una trombosis, el episodio agudo debe tratarse con heparina intravenosa y luego mantener anticoagulantes orales durante un tiempo. Pero aunque la mayoría aprueba el uso profiláctico, no se ha demostrado que sea beneficioso.
Se han hecho ensayos con globulina anti-timocito (dosis total de 150mg/kg de 4-10 días) y se ha demostrado que la terapia inmunosupresora parece beneficiar de forma selectiva a los pacientes con hipoproliferación (o sea, es efectiva en disminuir las citopenias periféricas), pero no se obtiene respuesta de disminución de la hemólisis (13).
Todos los pacientes que responden a esta terapia tienen como característica común una hipoproliferación, incluyendo las plaquetas (<30x109/l), y un grado menor de hemólisis crónica, representada por baja cantidad de reticulocitos (<100x109l), niveles de LDH (<1000 U/l) y la bilirrubina total (<17 mmol/l).
El factor estimulante de colonias de granulocitos (G-CSF) se está empleando para corregir la neutropenia severa con granulocitos del clon normal (14). Se puede intentar la doble terapia: G-CSF y ciclosporina (15). Esta combinación terapeútica se empleó en 4 pacientes con HPN que presentaban trombopenia severa, anemia hemolítica y neutropenia, observándose una respuesta favorable de la hematopoyesis en las 3 líneas celulares.
Actualmente se están haciendo estudios con la eritropoyetina recombinante humana (16) (pues, se supone que sólo estimula los clones normales de hematíes) y el ácido transretinoico (de forma muy limitada, ya que sólo se ha obtenido aumento del nivel de hemoglobina en patología asociada a trisomía 8) (17).
Por último, sólo mencionar los tratamientos que consisten en medidas de soporte: hierro, ácido fólico, transfusiones...
Bibliografía
1. Rosse WF. The natural history of paroxysmal nocturnal haemoglobinuria. Rev Invest Clin 1997; suppl 1, 8-9. [ Links ]
2. Rosse WF. Paroxysmal nocturnal hemoglobinuria as a molecular disease. Medicine 1997; 76:2,63-93. [ Links ]
3. Nishimura J, Smith CA, Phillips KL, Ware Re, Rosse WF. Paroxysmal nocturnal hemoglobinuria: molecular pathogenesis and molecular therapeutic approaches. Hematopathol Mol Hematol 1998; 11: 3-4,119-46. [ Links ]
4. Iwanga M, Furukawa K, Amenomori T, Mori H, Nakamura et al. Paroxysmal nocturnal haemoglobinuria clones in patients with myelodysplastic syndromes. Br J Haematol 1998; 102:2, 465-74. [ Links ]
5. Nishimura J, Inoue N, Wada H, Ueda E, Praoonjago P, Hirota T et al. A patient with paroxysmal nocturnal hemoglobinuria bearing four independent PIG-A mutant clones. Blood 1997; 89:9; 3470-6. [ Links ]
6. Ninomiya H, Hasegawa Y, Nagasawa T, Abe T. Excess soluble urokinase-type plasminogen activator receptor in the plasma of patients with paroxysmal nocturnal haemoglobinuria inhibits cell-associated fibrinolytic activity. Int J Hematol 1997; 65:3, 285-91. [ Links ]
7. Wiedmer T, Hall SE, Ortel Tl, Kane WH, Rosse WF, Simps PJ. Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood 1993; 82: 1192-1196. [ Links ]
8. Packman CH. Pathogenesis and management of paroxysmal nocturnal haemoglobinuria. Blood Rev 1998; 12:1, 1-11. [ Links ]
9. Iwanaga M, Furukaza K, Amenomori T, Mori H, Nakamura H, Fuchigami K et al. Paroxysmal nocturnal hemoglobinuria clones in patients with mielodysplastic syndromes. Br J Hematol 1998; 102:2,465-74. [ Links ]
10. Johnson RJ, Rawstron AC, Richards S, Morgan GJ, Norfolk DR, Hillmen SO. Circulating primitive stem cells in paroxysmal nocturnal hemoglobinuria (PNH) are predominantly normal in phenotype but granulocyte colony-stimulating factor treatment mobilizes mainly PNH stem cells. Blood 1998; 91:12,4504-8. [ Links ]
11. Rosse WF, Gutterman LA. The effect of iron therapy in paroxysmal nocturnal hemoglobinuria. Blood 1971; 36:559-565. [ Links ]
12. Harrington WJ Sr, Kolodny L, Horstman LL, J y W, Ahm YS. Danazol for paroxysmal nocturnal hemoglobinuria. Am JK Hematol 1997; 54:2, 149-54. [ Links ]
13. Paquette RL, Yoshimura R, Veiseh C, Kunkel L, Gajewski J, Rosen PJ. Clinical characteristics predict response to antithymocyte globulin in paroxysmal nocturnal haemoglobinuria. Br J Haematol 1997; 96:1, 92-7. [ Links ]
14. Je´go P, Le Strat A, Girard L, Sébillot M et al. Paroxysmal nocturnal hemoglobinuria: efficacy of prolonged treatment with granulocyte colony-stimulating factor. Blood 1997; 90:7,2841-3. [ Links ]
15. Schubert J, Scholz C, Geissler RG, Ganser A, Schmidt RE. G-CSF and cyclosporin induce an increase of normal cells in hypoplastic parowysmal nocturnal hemoglobinuria. Ann Hematol 1997; 74:5,225-30. [ Links ]
16. Astori C, Bonfichi M, Pagnucco G, Bernasconi P, Lazzarino M et al. Treatment with recombinant human erythropoietin in a patient with paroxysmal nocturnal haemoglobinuria: evaluation of membrane proteins CD55 an CD 59 with cytofluorometric assay. Br J Haematol 1997; 97: 3, 586-8. [ Links ]
17. Vinion N, Michali E, Meletis J, Andreopoulos A, Vaiopoulus G et al, Trisomy 8 in a patient who responded to therapy with all-trans-retinoic acid and developed paraxysmal nocturnal haemoglobinuria. Br J Haematol 1997; 97: 1,135-6. [ Links ]

