










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.22 no.10 oct. 2005
| Enfermedad de jarabe de arce: una entidad rara que debemos recordar. D. A. DE LUIS ROMÁN, O. IZAOLA JÁUREGUI Unidad de Apoyo a la Investigación. Instituto de Endocrinología y Nutrición. Facultad de Medicina.
|
| MAPPLE SYRUP DISEASE: A RARE ENTITY THAT WE MUST KNOW. REVIEW OF ITS DIETETIC MANAGEMENT
|
| RESUMEN La enfermedad de jarabe de arce o cetoaciduria de cadena ramificada es causada por una deficiencia en la actividad del complejo de la deshidrogenasa de los a cetoácidos de cadena ramificada. Esta enfermedad se hereda con carácter autósomico recesivo, afecta por igual a ambos sexos, siendo su incidencia de 1/200.000 recién nacidos. Basado en la presentación clínica y respuesta bioquímica a la administración de tiamina, estos pacientes se dividen en cinco fenotipos clínicos y bioquímicos diferentes: clásica, intermedia, intermitente, sensible a tiamina y deficiencia de dihidrolipoil deshidrogenasa (E3). En los pacientes con esta enfermedad se detecta un aumento cualitativo de aminoácidos ramificados en plasma, se puede apreciar por métodos utilizados en screening (cromatografía en capa fina) durante el periodo neonatal. Los aminoácidos valina, isoleucina, y aloisoleucina se encuentran aumentados en plasma, orina, y líquido cefalorraquídeo determinados por cromatografía de intercambio iónico, cromatografía de alta resolución o electroforesis alto voltaje. Es necesario diferenciar la fase en la que se encuentra el paciente (fase aguda o fase de mantenimiento). Los objetivos en la fase de descompensación metabólica aguda se basan en tres puntos: eliminar los metabolitos tóxicos, soporte nutricional y conseguir anabolismo. La utilización de exaguinotransfusión/hemodiálisis/diálisis peritoneal es una de las primeras medidas, junto con las modificaciones dietéticas que aportan un minimo energético y que intentan controlar los niveles de aminoácidos en sangre. La utilización de fórmulas dietéticas modificadas artificialmente constituye uno de los pilares del tratamiento en estos pacientes. PALABRAS CLAVE: Iodine. Deficiencia. Pregnancy. | ABSTRACT Mapple syrup disease is secondary to a deficiency of deshidrogenase complex of a cetoacid of branched-chain. This disease has a recesive autosomic inheritance, with an incidence of 1/200.000 newborns, without differences between male and female. Due to clinical presentation and biochemical response to tiamin, these patients can be classified in five clinical entities: classic, intermediate, intermittent, positive response to tiamin and defficience of dihidrolipoil deshidrogenase (E3). In these patients, an increase of seric branched-chain aminoacids is detected, it could be detected by (cromathography) during neonatal period. Valine, isoleucine, and aloisoleucine are incresaed in serum, orine, and cephaloraquideum liquid by ionic changed cromathography, cromathography of high resolution or high voltage electrophoresis. Patients have two phases in this disease (acute phase and mantaining phase). Objectives in acute phase are based in three topics: to eliminate toxic metabolites, nutritional support and to get anabolism. Utilization of hemodyalisis/peritoneal dyalisis/blood exchange is one of the first treatments. Dietetic support is the second treatment, with a minimun energy intake and controlling blood levels of aminoacids. Modified dietetic formulas is a main device to treat these patients. KEY WORDS: Iodine. Deficiency. Pregnancy.
|
De Luis Román DA, Izaola Jáuregui O. Enfermedad de jarabe de arce: una entidad rara que debemos recordar. A propósito de su manejo dietético. An Med Interna (Madrid) 2005; 22: 493-497.
Trabajo aceptado: 31 de mayo de 2005
Correspondencia: Daniel de Luis Román. C/ Los Perales, 16. 47130 Simancas. Valladolid. e-mail: dadluis@yahoo.es
INTRODUCCIÓN
La enfermedad de jarabe de arce o cetoaciduria de cadena ramificada es causada por una deficiencia en la actividad del complejo de la deshidrogenasa de los cetoácidos de cadena ramificada. Dicho complejo comprende tres componentes catalíticos: E1 o descarboxilasa con dos estructuras E1a y E1ß y dependiente de pirofosfato de tiamina, E2 o dihidrolipoil transacilasa y E3 o dihidrolipoil deshidrogenasa. Los estudios moleculares han permitido la localización en diferentes genes que codifican cada una de estas subunidades, ello explicaría la heterogenicidad genética de la enfermedad, así como los diferentes fenotipos moleculares en función del locus afectado del complejo deshidrogenasa de los a-cetoácidos (1).
Esta enfermedad se hereda con carácter autósomico recesivo, afecta por igual a ambos sexos, siendo su incidencia de 1/200.000 recién nacidos. La mayoría de los pacientes no tratados mueren por crisis recurrentes metabólicas y deterioro neurológico en los primeros días de vida. La edad del diagnóstico y el subsiguiente control metabólico son los datos más importantes determinantes para un buen pronóstico a lo largo del tiempo (2).
Este trastorno se caracteriza por un acúmulo de aminoácidos de cadena ramificada: leucina, isoleucina, valina y aloisoleucina y de los correspondientes a -cetoácidos de cadena ramificada: a-cetoisocaproico, a-ceto- ß-metilvalérico y a-cetoisovalérico. La presencia de aloisoleucina es patognomónica de la enfermedad.
El nombre de esta enfermedad se debe a que la orina tiene un olor característico a jarabe de arce.
MANIFESTACIONES CLÍNICAS
Basado en la presentación clínica y respuesta bioquímica a la administración de tiamina, estos pacientes se dividen en cinco fenotipos clínicos y bioquímicos diferentes: clásica, intermedia, intermitente, sensible a tiamina y deficiencia de dihidrolipoil deshidrogenasa (E3) (3,4).
FORMA CLÁSICA
Comienzo en los 3-4 primeros días de vida tras el inicio de la alimentación, con rechazo de la alimentación, letargia, alteraciones del tono muscular, convulsiones y coma. Analíticamente hay cetoacidosis con cetonuria, y puede existir hipoglucemia. Existe marcado aumento de los aminoácidos de cadena ramificada (AACR), especialmente leucina, y de los ácidos orgánicos (AAO) en los fluidos biológicos (plasma, líquido cefaloraquideo, orina), y presencia de aloisoleucina. La actividad enzimática en fibroblastos es menor del 2% con respecto a la actividad normal.
FORMA INTERMEDIA
Suele presentarse a partir del periodo lactante y a lo largo de la infancia, con los siguientes síntomas: retraso en el desarrollo pondero estatural y psicomotor, acompañado de convulsiones; puede existir oftalmoplejía en el periodo neonatal. Desde el punto de vista bioquímico se manifiesta por episodios de cetoacidosis y aumento persistente de AACR y AAO, aunque menos intenso que la forma clásica. La actividad enzimática residual es del 3 al 20% con respecto a lo normal.
FORMA INTERMITENTE
Aparece en los pacientes con desarrollo prácticamente normal, caracterizada como crisis de ataxia/cetoacidosis desencadenadas por procesos febriles y/o ingestas excesivas de proteínas. Cuando los pacientes se encuentran asintomáticos los niveles de AACR y AAO son normales. La actividad enzimática residual es del 5-20% con respecto a lo normal.
FORMA SENSIBLE A LA TIAMINA
No existe un criterio uniforme para su diagnóstico. En general, estos pacientes no tienen la enfermedad aguda y su curso clínico es parecido a la forma intermedia, caracterizándose por un aumento de BCAA, que se normaliza tras el tratamiento con dosis farmacológicas de tiamina, manteniendo una ingesta constante de proteínas. La actividad enzimática residual es del 2-40% con respecto a lo normal.
DEFICIENCIA DE DIHIDROLIPOIL DESHIDROGENASA (E3)
El componente E3 del complejo multienzimático es común para otros enzimas como piruvato deshidrogenasa y a -cetoglutamato deshidrogenasa, es por lo que en pacientes con enfermedad de jarabe de arce por déficit en E3 también se produce acidosis láctica y a -cetoglutárica, además de un incremento de a-cetoácidos de cadena ramificada. El fenotipo clínico es similar a la forma intermedia de la enfermedad, pero acompañada de un aumento de láctico, pirúvico, a -cetoglutarato, a -hidroxivalérico, y a -hidroxiglutárico. Los AACR están moderadamente aumentados en plasma en comparación con la forma clásica. Los pacientes presentan deficiencia combinada del complejo enzimático y de los complejos piruvato y a -cetoglutarato deshidrogenasas.
FORMAS NO CLASIFICABLES
Existen formas con alelos mutantes que se comportan como heterocigotos.
APROXIMACIÓN DIAGNÓSTICA
En los pacientes con esta enfermedad se detecta un aumento cualitativo de aminoácidos ramificados en plasma, se puede apreciar por métodos utilizados en screening (cromatografía en capa fina) durante el periodo neonatal. Los aminoácidos valina, isoleucina, y aloisoleucina se encuentran aumentados en plasma, orina, y líquido cefalorraquídeo determinados por cromatografía de intercambio iónico, cromatografía de alta resolución o electroforesis alto voltaje (5). Con un perfil normal de estos aminoácidos no se puede excluir el diagnóstico ya en la forma intermitente solamente se acumulan en los episodios agudos.
Es necesario tener en cuenta que la presencia de aloisoleucina es patognomónica de esta enfermedad. La formación aloisoleucina sucede por racemización de L-isoleucina, tautomerización y transaminación del a-ceto a-metilisovalérico. Este metabolito persiste elevado varios días tras un episodio de descompensación.
En diagnóstico bioquímico es útil la determinación de ácidos orgánicos. El método de 2,4-dinitrofenilhidracina es una prueba simple, la presencia de a-cetoácidos aumentados en orina, produce un precipitado amarillo. Sin embargo es con el análisis de ácidos orgánicos en plasma, suero y orina por cromatografía de gases/espectrometría de masas en el que se obtiene un perfil característico de la enfermedad. Los a-cetoácidos presentes se convierten a trimetilsilil ésteres, apareciendo un espectro característico de los derivados trimetisil de a-cetoisovalérico, a-cetoisocaproico y a-ceto-metilvalérico. El a-hidroxivalérico, que se deriva del a-cetoisovalérico, es el metabolito mayoritario y de valor diagnóstico en estos enfermos. En pacientes con la forma intermitente el perfil de los a-cetoácidos es normal durante la remisión, por lo tanto el diagnóstico se ha de hacer cuando el paciente presenta síntomas.
Recientemente se ha utilizado la técnica de bombardeo rápido de átomos acoplado a la espectrometría para investigar a la vez trastornos de metabolismo de aminoácidos y ácidos orgánicos en muestras de sangre y orina impregnados en papel (5). Esta técnica podría permitir en los próximos años el análisis definitivo en muestra impregnada de sangre de talón para diagnóstico de esta patología.
Dada la linealidad entre los aminoácidos valina, leucina e isoleucina y sus respectivos a-cetoácidos, la monitorización de los pacientes se puede llevar a cabo teniendo en cuenta los niveles plasmáticos de los aminoácidos de cadena ramificada en plasma.
El diagnóstico neonatal se realiza mediante la determinación de la deficiencia en la descarboxilación de [1-C14] leucina, isoleucina y valina, en leucitos y cultivos de fibroblastos de los pacientes. El diagnóstico prenatal se realiza mediante análisis directo en tejido o en cultivos celulares de vellosidades coriónicas incubados con [1-C14] leucina como sustrato.
TRATAMIENTO
Es necesario diferenciar la fase en la que se encuentra el paciente (fase aguda o fase de mantenimiento) (6). Los objetivos en la fase de descompensación metabólica aguda se basan en tres puntos: eliminar los metabolitos tóxicos, soporte nutricional y conseguir anabolismo.
El primer aspecto se consigue con diálisis peritoneal, hemodiálisis o exanguinotransfusión, siendo desde el punto de vista práctico la diálisis peritoneal la más efectiva, puesto que la hemodiálisis aunque reduce más rápidamente los niveles de AACR, presenta problemas técnicos en los lactantes pequeños, y la exanguinotransfusión, que también consigue descensos significativos de los AACR, vuelven a incrementarse rápidamente tras su finalización (7). En cualquier caso, la elección de una u otra técnica depende de las disponibilidades y experiencia de cada hospital. La técnica escogida debe iniciarse con urgencia en pacientes metabólicamente descompensados, aunque existe controversia sobre las indicaciones precisas. Es fundamental inducir anabolismo con fórmulas alimentarias exentas de AACR, o con nutrición parentenal si su estado es crítico. Se acepta que las indicaciones de iniciar al mismo tiempo diálisis peritoneal o hemodiálisis son: la no tolerancia de la alimentación por sonda nasogástrica, concentración de leucina plasmática superior a 2.500-3.000 nmol/l (32-39 mg/dl), síntomas neurológicos graves, edad mayor de 11-15 días de vida; así mismo, debe iniciarse una de estas técnicas si tras 24 horas de nutrición oral/parentenal hay deterioro clínico y/o concentración de leucina ha descendido menos de 500 nmol/L (6,5 md/dl), permaneciendo por encima de 1000 nmol/L (13 mg/dl).
La nutrición parentenal debe realizarse con una mezcla de aminoácidos exenta de AACR con alto contenido energético en forma de glucosa (10-29 mg/kg/minuto), aporte de líquidos adecuados (130-190 ml/kg/día), añadiendo si se precisa insulina en cantidad suficiente para mantener glucemias entre 100-130 mg/dl. La administración de lípidos hasta 2,5 mg/kg/día permite aumentar el aporte energético (8).
En la fase de mantenimiento, el objetivo del tratamiento es mantener las concentraciones plasmáticas de los AACR en los niveles próximo posible a los valores normales. La leucina es el AACR que se encuentra en mayor proporción en los alimentos naturales (Tabla I) y es el más neurotóxico, por lo que su estricta monitorización es fundamental, debiéndose mantener su concentración plasmática entre 2-6 mg/dl.
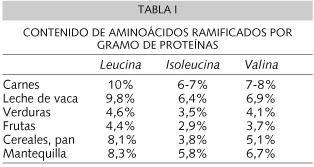
Los AACR son aminoácidos esenciales, aunque sus requerimientos no están exactamente definidos. Las RDA recomiendan los siguientes aportes desde el primer semestre de vida hasta los 10 años: leucina 161 a 42 mg/kg, isoleucina 70 a 28 mg/kg y valina 93 a 25 mg/kg. Sin embargo, existen numerosas diferencias individuales, por lo que su aporte debe ser pautado dependiendo de sus concentraciones plasmáticas (9-10). Los aportes de leucina suelen ser estables entre 300-600 mg/día durante los primeros cinco años de vida (11,12).
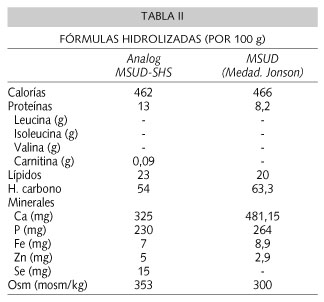
Tras superar la fase de descompensación debe iniciarse el aporte de proteínas con fórmulas hidrolizadas exentas de AACR (Tabla II), y aportar éstos con una fórmula de inicio, mezcladas ambas fórmulas. Se recomienda un aporte total de aminoácidos o equivalente proteico de 3 g/kg/día (0,5-1,7 g/kg/día de proteínas naturales y el resto mediante la fórmula hidrolizada de AACR). En ocasiones es muy difícil conseguir que las concentraciones de los tres AACR se mantengan dentro de los límites deseables, puesto que si se consigue mantener la leucina por debajo de 6 mg/dl, las concentraciones de isoleucina y valina pueden ser inferiores a 1,5-2 mg/dl, en cuyo caso hay que suplementar la mezcla de las fórmulas (exenta de AACR y de inicio) con el aminoácido deficitario (dosis 200 mg/kg/día) hasta conseguir su normalización.
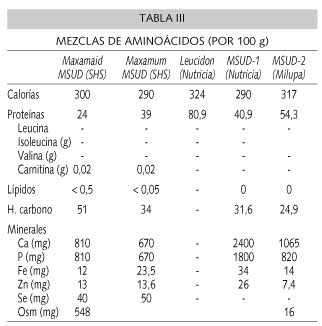
En niños mayores y adolescentes el aporte principal de proteínas se realiza con mezclas de aminoácidos exentos de AACR (Tabla III), y aportando éstos con alimentos de bajo contenido proteico, como verduras, hortalizas y frutas (Tabla I). Para alcanzar las necesidades calóricas se añaden alimentos libres, totalmente exentos de AACR, como aceites (oliva, maíz, girasol, MCT y dextrinomaltosa). Algunos pacientes toleran alto aporte de proteínas naturales, entre 6-10 g/día, que pueden ser administradas mediante alimentos con proteínas de alto valor biológico.
Hay que realizar controles analíticos nutricionales para prevenir y/o corregir posibles deficiencias (13,14), habiendo sido descritas anemias megaloblásticas por deficiencia de ácido fólico, deficiencia de selenio, carnitina, etc. Se recomienda suplemento de por lo menos 50 mg/día de tiamina, aunque deben tratarse según tolerancia individual de cada paciente y dosis farmacolológicas de tiamina, aunque estas dosis oscilan en un amplio margen de 100 a 1.000 mg/día.
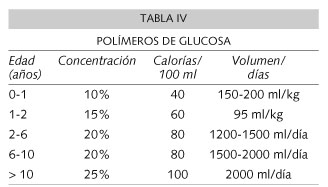
Durante los episodios febriles existe un gran riesgo de descompensación. Inicialmente los análisis más rutinarios, como equilibrio ácido-base, glucosa, ácido láctico y amonio suelen ser normales y puede no haber cetosis; sólo se alteran precozmente las concentraciones de AACR de cuyos valores no suele disponerse con urgencia. Por ello es fundamental objetivar síntomas sutiles como somnolencia, decaimiento y cambios de carácter, para sospechar el inicio de una crisis de descompensación. Los padres deben ser advertidos de estos hechos y enseñados para dar precozmente una solución de polímeros de glucosa, para evitar el catabolismo proteico, que tiene las ventajas de poder ser administrado por boca de fácil preparación y que no suele ser rechazado por el paciente por su sabor. En la tabla IV, se muestra la concentración y volumen a administrar, dependiendo de la edad, debiéndose dar inicialmente con mucha frecuencia, p. ej. 10 ml cada 10-15 minutos y si se tolera pasar a tomar cada 1-2 horas, o bien administrar por sonda nasogástrica.
PRONÓSTICO
En la forma clásica de la enfermedad el pronóstico es malo, ya que puede evolucionar con múltiples episodios de cetoacidosis desencadenados por infecciones, vacunaciones, estrés y ayunos prolongados (15). Dicho pronóstico depende de varios factores que deben ser evaluados conjuntamente: edad de diagnóstico, concentración de leucina a dicha edad, concentración media de leucina durante los primeros años, etc. Con tratamientos eficaces iniciados los primeros 10-20 días de vida se consigue desarrollo psicomotor normal a los 3-5 años de edad. Las formas intermedias, intermitentes y sensibles a tiamina tienen mejor pronóstico. Existe en la actualidad experiencia muy limitada en el transplante hepático (16), aunque en un futuro próximo constituirá una alternativa al tratamiento, igual que la terapia génica.
Bibliografía
1. Raimann E, Vallejos M, Pizarro T, Rodríguez L Proyecto conjunto de desarrollo y control de las enfermedades metabólicas. Hospital Roberto del Río e Instituto de Nutrición y Tecnología de los alimentos. Rev Pediatría (Santiago) 2002; 45: 74-75. [ Links ]
2. Cornejo V, Raimann E. Errores innatos del metabolismo de los aminoácidos. Capítulo 3, en Errores en el metabolismo del niño, editoras, Colombo M, Cornejo V, Raimann E. Editorial Mediterráneo, 1999. p. 59-106. [ Links ]
3. Fernández Sánchez A, Dalmau Serra J, GarcÍa Gómez AM, Cabello Tomás ML, Martínez Pardo M. Protocolo de diagnóstico y tratamiento de la enfermedad de jarabe de arce. An Esp Pediatr 1997; 89: 9-13. [ Links ]
4. Cidras Pidre M, García Martínez R, Feret Siguile MA, Clemente Yago F, Escriva Tomás P, Orts Serrano F et al. Enfermedad de jarabe de arce: un caso diagnóstico precoz y tratamiento dietético. An Esp Pediatr 1991; 34: 92-94. [ Links ]
5. Deng C, Li N, Zhang X. Rapad determination of amino acids in neonatal blood simples based on derivatization with isobutyl chloroformate followed by solid-phase microextraction and gas chromatography mass spectrometry. Rapid Commun Spectrom 2004; 18: 2558-2264. [ Links ]
6. Colombo M, Raimann E, Cornejo V. Tratamiento de errores congénitos del metabolismo. En: Libro de apuntes Médicos. III Jornadas de Terapéutica en Pediatría. Editor Dr. J. Infante, 1990, p. 17-18. [ Links ]
7. Lin MC, Chen CH, Fu LS, Jan SL. Management of acute decompensation of neonatal maple syrup urine disease with continuos arteriovenous haemofiltration: report of one case. Acta Paediatr Taiwan 2002; 43: 281-284. [ Links ]
8. Jardim LB, Martins CS, Pires RF, Sanseverino MT, Refosco L, Viera R de C. Management of a case of maple syrup urine disease- the use of glucoinsulinotherapy. J Pediatr 1995; 71: 279-284. [ Links ]
9. L. Kathleen Mahan. Sylvia Escott-Stump. Nutrición dietoterapia de Krause. Décima Ed. Mc Graw Hill 2001, p. 1065-1083. [ Links ]
10. Riazi R, Rafii M, Clarke JT, Wykes LJ. Total branched-chain amino acids requirement in patients with maple syrup urine disease by use of indicator amino acid oxidation with L-(1-13C). Am J Physiol Endocrinol Metab 2004; 287: 142-194. [ Links ]
11. Durán G, Raiann E, Mabe P, Cornejo V, Jiménez M, De la Parra A, et al. Experience in treatment of organic acidurias in Chile. J Inherit Metab Dis 2000; 23: 85. [ Links ]
12. Cabello JF, Cornejo V, Raimann E, Colombo M. Experience in treatment of organic acidurias in Chile. J Inherit Metab Dis 2000; 23: 81. [ Links ]
13. Nyhan WL, Rice-Kelts M, Klein J, Barshop BA. Treatment of the acute crisis in maple syrup urine disease. Arch Pediatr Adolesc Med 1998; 152: 593-598. [ Links ]
14. Durán G, Cornejo V, Valiente A, Muñoz L, Raimann E. Carnitine status in Phenylketonuric patiens on dietary treatment in Chile J Inherit Metab Dis 2000; 23: 24. [ Links ]
15. Chuag DT.Maple syrup urine disease: it has come a long way. J Pediatr 1998; 132S: 17-23. [ Links ]
16. Wendel U, Saudubray JM, Bodner A, Schadewaldt P. Liver transplantation in maple syrup urine disease. Eur J Pediatr 1999; 158: 60-64. [ Links ]

