










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAvances en Odontoestomatología
versión On-line ISSN 2340-3152versión impresa ISSN 0213-1285
Av Odontoestomatol vol.20 no.5 Madrid sep./oct. 2004
Hemostasia y tratamiento odontológico
Quintero Parada E*, Sabater Recolons MM**, Chimenos Kustner E***, López López J***
|
INTRODUCCIÓN
Actualmente la valoración de un paciente con historia de hematomas y hemorragias es un problema clínico frecuente. El diagnóstico y el tratamiento adecuado de estos pacientes dependen del conocimiento profundo de los mecanismos normales de la hemostasia y de los análisis que estudian estos mecanismos (1). Las manipulaciones o tratamientos que se realizan en la cavidad bucal, especialmente aquellos que puedan ocasionar la extravasación de sangre, constituyen un riesgo importante para los pacientes con trastornos de la hemostasia. La mejor forma de evitar complicaciones hemorrágicas tras procedimientos quirúrgicos bucales es siempre la prevención y para ello es indispensable disponer de una historia clínica detallada del paciente, haciendo especial énfasis en los antecedentes de problemas hemorrágicos tras alguna intervención, parto o extracción dentaria y también en cuanto a antecedentes médicos tanto personales como familiares, que pudieran influir en una correcta hemostasia (2). Es importante recordar que la mayoría de hemorragias menores, posteriores a una cirugía bucal, suelen estar en relación con factores locales dependientes del entorno anatómico. Sin embargo, la mayor parte de hemorragias graves tras un tratamiento bucal quirúrgico se relacionan con trastornos sistémicos de la hemostasia, tales como hepatopatías primarias o hipertensión arterial, entre otras (2).
La hemostasia es un mecanismo constituido por varios sistemas biológicos interdependientes, cuya finalidad es conservar la integridad y permeabilidad del sistema circulatorio; es decir, que el término hemostasia significa prevención de la pérdida de sangre (3, 4).
Siempre que se lesiona o se rompe un vaso, la hemostasia se consigue mediante diversos mecanismos:
1. El espasmo vascular. Inmediatamente después de que se lesiona o se rompe un vaso, el traumatismo de su pared provoca su contracción y reduce el flujo de sangre procedente del vaso roto (4, 5).
2. La formación de un agregado o trombo de plaquetas sobre la superficie vascular lesionada. Las plaquetas constituyen el trombo plaquetario, el cual proporciona la hemostasia primaria o provisional, y también intervienen en la coagulación plasmática. Las plaquetas se adhieren a las estructuras subendoteliales que han quedado expuestas por la lesión. Las plaquetas producen serotonina y tromboxano A2 que realizan tres funciones: aumentar la adhesión plaquetaria iniciada, aumentar la vasoconstricción del vaso o vasos sanguíneos y por último contribuir a la activación de los factores de la coagulación X y II. Dependiendo de la magnitud de la rotura del vaso, las plaquetas requieren una proteína plasmática, denominada factor de Von Willebrand, que le permite su adhesión a la matriz endotelial subepitelial expuesta. La adhesión de estas plaquetas en la zona de la lesión vascular va seguida rápidamente por la agregación de grandes cifras de plaquetas para formar el tapón plaquetario, completándose así la hemostasia primaria (1, 5, 6).
3. La formación de fibrina. El tercer mecanismo de la hemostasia es la formación del coágulo de sangre. La coagulación plasmática o formación de fibrina consiste en la trasformación del fibrinógeno (soluble) en fibrina (insoluble), por medio de la trombina, la cual es una enzima proteolítica que se forma por activación de la protrombina. La protrombina y el fibrinógeno, junto a otras proteínas, constituyen los factores de coagulación necesarios para la formación de fibrina. La coagulación intensifica la hemostasia iniciada con la vasoconstricción y desarrollada por las plaquetas. Estos factores de coagulación son proteínas, de las que se distinguen tres grupos: factores dependientes de la vitamina K, factores sensibles a la trombina y factores de contacto (6). La transformación de protrombina en trombina se considera que ocurre por dos vías, aunque en realidad éstas interactúan constantemente, las cuales están representadas en la tabla I (7).

4. La eliminación de los depósitos de fibrina o fibrinólisis. Este proceso destruye la fibrina formada durante la coagulación. Se caracteriza por la activación de la plasmina a partir de un precursor inactivo del plasma, el plasminógeno. La acción impulsora que ejerce la trombina sobre la hemostasia se ve limitada por la misma trombina, actuando como un seguro, que evita que la hemostasia vaya más lejos del hecho de restablecer el vaso dañado, prolongándose en el tiempo. Esta acción limitadora la realiza la trombina activando un receptor que se encuentra a nivel de la membrana endotelial que se denomina trombomodulina. Desde el momento que la trombina se une a este receptor se produce la denominada proteína e, que es un potente inhibidor de la coagulación (6, 8, 9).
Los factores que intervienen en la coagulación y otras denominaciones con las que se correlacionan se pueden apreciar en la tabla II (6).

El propósito de esta revisión bibliográfica es desarrollar en el área de las coagulopatías el tratamiento y manejo odontológico de las alteraciones que se pueden encontrar con mayor frecuencia en el gabinete odontológico.
CLASIFICACIÓN DE LOS TRASTORNOS DE LA HEMOSTASIA
En el campo de la hematología se produce una gran variedad de enfermedades que incluyen no sólo los trastornos de los órganos hematopoyéticos y los órganos celulares de la sangre periférica, sino también alteraciones de los sistemas Iinforeticular y hemostático. Este último representa una compleja interacción entre los vasos sanguíneos, las plaquetas y los factores plasmáticos de la coagulación (10). Las alteraciones de la hemostasia se clasifican según la fase de la coagulación que se encuentre afectada: trastornos de la hemostasia primaria (encargada de formar el tapón plaquetario) (11) y trastornos de la hemostasia secundaria (depende de las proteínas plasmáticas y de los factores de la coagulación) (2,6) (Tabla III).

Existen una serie de datos que a grandes rasgos, pueden ayudar a diferenciar si un trastorno de la hemostasia es de origen 1ª o 2° y se resumen a continuación:
Si la hemorragia que aparece tras un traumatismo o manipulación quirúrgica es inmediata, se tratará de una alteración de la hemostasia primaria. En ésta la hemorragia suele ser superficial (mucosa oral, piel, nariz) y en la clínica se manifiesta en petequias, equimosis o lesiones purpúricas. Si la hemorragia aparece al cabo de unas horas o días, orientará hacia una alteración de la hemostasia secundaria, que se caracteriza por afectar zonas profundas (articulaciones, músculos o espacios retroperitoneales), y la clínica se manifiesta en forma de hematoma. En cuanto al tratamiento, en general, en las alteraciones de la hemostasia primaria es suficiente con medidas locales, ya que la respuesta suele ser inmediata. No ocurre igual en los trastornos de la hemostasia secundaria, que suelen requerir un tratamiento sistémico mantenido (2). Las pruebas de valoración y detección, tanto de la hemostasia primaria como secundaria, se explican en la Tabla IV.

Con frecuencia, las enfermedades hematológicas se acompañan de lesiones orales significativas, pudiendo éstas constituir la primera manifestación clínica de las mismas. Las repercusiones en la cavidad bucal de muchas enfermedades hematológicas son prácticamente constantes. Diversos signos a nivel de la mucosa oral, tales como palidez, petequias y equimosis, uIceraciones, hipertrofia gingival, gingivorragias, etc., pueden deberse a un trastorno hematológico existente (12).
A continuación se detallan algunos aspectos de las coagulopatías más comunes y su manejo odontológico.
TELANGECTASIA HEMORRÁGICA HEREDITARIA O ENFERMEDAD DE RENDU-OSLER
Es un raro trastorno mucocutáneo que se hereda de forma autosómica dominante. Se caracteriza por la presencia de numerosos hamartomas vasculares (pequeñas colecciones de capilares dilatados), que afectan a la piel y a las mucosas, por lo que se considera una angiopatía neoformativa de telangiectasias circunscritas, que, al romperse determinan síndromes hemorrágicos locales (13, 14).
Manifestaciones clínicas
Se caracteriza por la presencia desde el nacimiento de múltiples telangiectasias en piel y mucosas. Son pacientes propensos a hemorragias localizadas, principalmente nasales (en forma de epistaxis), urinarias y con menor frecuencia digestivas y respiratorias (6, 15). El aspecto macroscópico de las lesiones es el de unas manchas de color rojo vinoso, cuyo tamaño varía entre el de una cabeza de alfiler y el de una lenteja, las cuales, al ser comprimidas (vitropresión), desaparecen casi por completo. Estos angiomas se presentan siempre en forma múltiple y se localizan principalmente en frente, pómulos, nariz, mentón, lengua, paladar y faringe. Generalmente estos pacientes presentan anemia ferropénica, por el sangrado de las telangiectasias gastrointestinales (6).
El diagnóstico clínico se basa en cuatro criterios: historia familiar, epistaxis, telangiectasias mucocutáneas y malformaciones arteriovenosas. Se realiza un diagnóstico definitivo si están presentes tres de los criterios mencionados (15).
Manifestaciones orales
Las lesiones orales se localizan en la piel alrededor de los labios y en la lengua, encía, mucosa bucal y con menos frecuencia en el paladar. (1, 14).
Tratamiento
Las medidas antihemorrágicas son sólo sintomáticas, presión local, aplicación de trombina. Las tela ngiectasias con hemorragia copiosa deben cauterizarse con nitrato de plata o ácido crómico (6).
Manejo odontológico
Las hemorragias se pueden controlar con preslon local o con agentes hemostáticos. En algún tratamiento protésico o periodontal puede ser necesaria la exéresis de estas lesiones. El láser de CO2 se indica como el tratamiento ideal para los tumores vasculares, ya que al disminuir considerablemente la hemorragia se evita el riesgo de aspiración así como la necesidad de transfusiones (16). Se recomienda el uso profiláctico de antibióticos, debido al 1% de prevalencia de abscesos cerebrales. La profilaxis que se recomienda es la misma que para la endocarditis bacteriana (13).
PÚRPURA TROMBOCITOPÉNICA IDIOPÁTICA (PTI)
La PTI es una enfermedad relativamente frecuente, en la que se produce una alteración de plaquetas aislada en individuos sanos.
Manifestaciones clínicas
Se manifiesta bajo dos formas: aguda y crónica. La PTI aguda se presenta con más frecuencia en niños, aunque puede ocurrir a cualquier edad. Se caracteriza por un inicio súbito con trombocitopenia, que da lugar a hematomas, hemorragias y petequias, pocos días o semanas después de una enfermedad vÍrica. Suele ser un proceso autolimitado, que remite, en general sin secuelas. La PTI crónica es más frecuente en adultos y su inicio puede ser súbito o insidioso. Es tres veces más común en mujeres que en hombres y su etiología es desconocida, pero el mecanismo patogénico es una destrucción periférica de las plaquetas, de base inmunológica con la detección de anticuerpos antiplaquetarios. La clínica deriva del déficit plaquetario, por lo que consiste en hematomas y hemorragias de gravedad variable, así como petequias y equimosis.
Manifestaciones orales
En muchas ocasiones las manifestaciones orales de la trombocitopenia suelen ser el primer signo de la enfermedad y clínicamente se pueden observar como petequias, equimosis o vesículas hemorrágicas de localización variable, principalmente en la lengua, los labios y en la línea oclusal o línea alba, secundarias a pequeños traumatismos. También son frecuentes pequeñas hemorragias tras cepillado, exodoncias de dientes temporales o tratamiento periodontal, así como espontáneas. El diagnóstico es básicamente clínico y de exclusión. Entre los datos de laboratorio sólo se encuentra un trastorno plaquetario.
Tratamiento
El tratamiento se centra principalmente en la administración de corticoides y/o la esplenectomía (1, 2).
Manejo odontológico
Las hemorragias gingivales espontáneas pueden tratarse generalmente con enjuagues oxidantes, pero pueden requerir transfusiones plaquetarias para detener la hemorragia. Una buena higiene oral y el tratamiento periodontal conservador ayudan a eliminar la placa y el cálculo que potencian las hemorragias. Para el tratamiento dental son deseables niveles plaquetarios mayores de 50.000/mm3. El tratamiento de urgencia durante los episodios trombocitopénicos graves consiste en el tratamiento endodóntico, antibiótico y analgésico evitando los AINES que impidan la agregación plaquetaria, de tipo no-salicilato. Las hemorragias postoperatorias se pueden controlar con colágeno microfibrilar (1, 2).
HEMOFILIA
La hemofilia es un desorden ligado al cromosoma X, que ocurre en 1 de 5.000 varones nacidos. El grado de déficit del factor se correlaciona con la frecuencia y significancia del sangrado. Existen dos tipos de hemofilia: la hemofilia A y la B (17).
HEMOFILIA A
Define una anomalía congénita del factor VIII de la coagulación sanguínea. El tipo de herencia de la hemofilia A es recesiva, ligada al cromosoma X. Este hecho determina que la hemofilia A se manifieste en varones, mientras que las mujeres son portadoras de la enfermedad (6).
Manifestaciones clínicas
Sólo de forma esporádica aparecen hemorragias en las mucosas. La frecuencia y la intensidad de las manifestaciones hemorrágicas guardan relación con las concentraciones del factor VIII circulante. Cuando éstas son indetectables, la hemofilia se clasifica como grave. Concentraciones inferiores al 5% definen la forma moderada, y cifras entre el 5 y el 50% corresponden a la forma leve (6). En las formas graves los pacientes presentan hemorragias espontáneas, en las articulaciones y a veces en los tejidos blandos, y cualquier traumatismo menor puede asociarse con hemorragias de peligro vital. Las diátesis hemorrágicas afectan sobre todo a las articulaciones, los músculos, el sistema genitourinario y el SNC. Las hemartrosis son dolorosas, incluso antes de aparecer signos clínicos. Los pacientes con enfermedad leve rara vez presentan episodios hemorrágicos, excepto cuando son sometidos a cirugías o sufren traumatismos. En la hemofilia moderada pueden producirse hemorragias espontáneas con poca frecuencia, pero puede requerir tratamiento por traumatismos menores varias veces al año (1, 2).
El diagnóstico se suele realizar en la historia clínica. Se puede sospechar una hemofilia por antecedentes hemorrágicos familiares de presentación exclusiva en varones, por la respuesta exagerada a pequeños traumatismos o manipulaciones quirúrgicas, o por el hallazgo en pruebas de hemostasia sistemáticas, de una prolongación del tiempo de tromboplastina parcial activado (TTPA), acompañados de tiempo de hemorragia y protrombina normales. El diagnóstico definitivo se establece al comprobar el descenso o la ausencia de la actividad procoagulante del factor VIII plasmático (6).
Manifestaciones orales
La presentación oral más frecuente son las hemorragias gingivales episódicas, prolongadas, espontáneas o traumáticas. Puede producirse, aunque es poco frecuente, hemartrosis de la articulación temporomandibular. También se pueden encontrar los llamados seudotumores de la hemofilia, que consisten en inflamaciones quísticas progresivas producidas por hemorragias recurrentes, que pueden acompañarse de signos radiológicos de afectación ósea (1). En estudios realizados por Sonbol et al. se evaluó la presencia de placa bacteriana y el índice de caries en pacientes con hemofilia, comparado con un grupo control, y se concluyó que el índice de caries en pacientes con hemofilia es significativa mente menor comparado con niños saludables, al igual que el índice de placa bacteriana, que también fue menor (18).
Tratamiento
El tratamiento de elección en la hemofilia grave es la utilización de preparados del factor VIII. Es conveniente tener siempre presente que el tratamiento de los episodios hemorrágicos debe iniciarse lo más rápido posible, ya que puede limitar las lesiones residuales. El crioprecipitado fue la primera medida eficaz, pero no cumplía con los requisitos de inactivación vírica, por lo que su uso ha sido desplazado por los concentrados plasmáticos sometidos a procedimientos fisicoquímicos de inactivación vírica, y así se disminuye el riesgo de transmisión de agentes infecciosos. Para establecer las dosis necesarias para un episodio hemorrágico se toman en cuenta dos parámetros: el peso del paciente y la dosis de factor VIII que se desea alcanzar. En estos pacientes se puede controlar la hemorragia administrando el factor ausente, hasta un nivel proporcional a la agresión que se va a realizar, así como un tratamiento antifibrinolítico. La sutura se debe mantener un mínimo de 10 días (2, 6).
La administración repetida de hemoderivados tiene el riesgo potencial de inducir la aparición de complicaciones:
- De carácter infeccioso.
- Desarrollar un inhibidor contra el factor VIII.
- La administración repetida de proteínas presentes en los concentrados plasmáticos puede generar un estado de inmunopresión (6).
Dado que el 20-30% de los hemofílicos tratados desarrollan un inhibidor contra el factor VIII, la detección de este inhibidor circulante puede complicar notablemente el tratamiento de estos enfermos. En esos pacientes se han utilizado pautas terapéuticas muy variadas, incluyendo factor VIII de origen porcino y altas dosis de inmunoglobulinas, aunque esto es de utilidad sólo en pacientes con bajos título de inhibidores. Recientemente se ha estudiado una terapia mediante un complejo coagulante antiinhibidor, para rescatar la terapia de concentrados de factor VIII, en pacientes que han desarrollado inhibidores de factor VIII. El compuesto contiene porciones de factor VIII y recombinante humano del factor VIIa, conseguido por recombinación genética (2, 6, 19). Recientemente se administró este complejo (Autoplex T), a un paciente que había desarrollado inhibidores del factor VIII, estabilizándose la hematuria y la hemartrosis; en un período de 6 meses no se ha produjo sangrado adicional desde la última dosis del complejo (19). Esto es de utilidad en complicaciones hemorrágicas en pacientes con altos títulos de inhibidor (6).
Es importante considerar la prevención y el control de la neutralización de anticuerpos e infecciones por hepatitis, así como VIH, en los concentrados plasmáticos del factor a reponer, para así mejorar sustancialmente la longevidad para personas con hemofilia. Además se ha asociado un incremento del riesgo relacionado a padecer hepatitis C y enfermedad hepática en personas con hemofilia, particularmente por infección por virus de la hepatitis B, con bajos niveles de Iinfocitos CD4 o pacientes ancianos (20, 21).
HEMOFILIA B
También llamada enfermedad de Christmas, es una coagulopatía congénita secundaria a una anomalía cuantitativa o cualitativa del factor IX de la coagulación. Al igual que en la hemofilia A, el gen que codifica al factor IX se encuentra en el cromosoma X, lo que la convierte en enfermedad ligada al sexo (1, 6).
Manifestaciones clínicas
Las manifestaciones de la enfermedad de Christmas son idénticas a las de la hemofilia A, con los mismos problemas musculoesqueléticos y las mismas complicaciones derivadas del tratamiento crónico con transfusiones. La gravedad del cuadro hemorrágico está relacionada con la actividad funcional del factor IX circulante, por lo que los criterios de clasificación de la enfermedad son los referidos para la hemofilia A (1,6).
El diagnóstico del déficit del factor IX es muy evidente. El tiempo de tromboplastina parcial (TTP) está prolongado, el tiempo de protrombina (TP) y el tiempo de sangría son normales, y los análisis de factores específicos confirman el diagnóstico. Las manifestaciones orales se correlacionan con las de la hemofilia A (1).
Tratamiento
Es similar al señalado para la hemofilia A. En este caso, los productos que se han de utilizar son los concentrados plasmáticos, previamente sometidos a inactivación vírica, de complejo protrombínico (mezcla de factores II, VII, IX y X) o concentrados específicos de factor IX. El uso de 25-50 U/kg de factor IX suele ser suficiente para alcanzar una buena hemostasia; se aconsejan perfusiones del preparado cada 12 o 24 h (6).
Manejo odontológico de las hemofilias
- Es importante que el odontólogo y estomatólogo conozcan la enfermedad y sus complicaciones, además deben ser capaces de explicar al paciente cómo un buen cuidado dental minimiza el riesgo de la intervención.
- El tratamiento dental debe ir dirigido a la prevención, comenzando por la educación tanto del paciente como de los padres en relación con el cuidado dental. Una buena higiene oral ayuda a reducir la hemorragia gingival. El uso de fluoruros, sellantes de fisuras, consejos en la dieta para restringir el consumo de azúcar y una inspección dental regular a edades tempranas son medidas importantes que favorecerán la vida del diente en boca. Prevenir la enfermedad periodontal es también importante (1, 22).
- En el caso de que ya no se pueda realizar una pauta preventiva, sino que haya que realizar tratamientos invasivos, debe considerarse la estabilidad y el control de la enfermedad. Es decir, el paciente debe tener su pauta de reposición de factor deficitario, dependiendo del tratamiento a realizar, por lo que es indispensable consultar con el hematólogo. Para prevenir los hematomas en la realización de una radiografía dental, o por el uso de aspiradores, se puede colocar espuma en la punta de la goma o una gasa en los instrumentos.
- La mayor parte de los tratamientos conservadores se pueden realizar sin reposición del factor, aunque se recomienda el uso del dique de goma para prevenir lesiones de los tejidos blandos.
- Los dientes primarios deben ser extraídos en cuanto comiencen a moverse. Cuando la radiografía revela sólo unión al tejido blando, hay que plantear un programa de higiene, dos días antes de hacer la extracción. La hemorragia inicial se puede controlar con presión o medidas hemostáticas, como trombina o colágeno fibrilar (1). La cirugía oral en pacientes con hemofilia requiere un manejo especial en colaboración con el hematólogo.
- La cirugía oral menor de los casos no complicados y que tengan niveles estables del factor ausente, puede ser realizada por el dentista o un cirujano oral, con la ayuda y asistencia del hematólogo. Las cirugías mayores en pacientes que tengan inhibidores contra el factor VIII, deben ser realizadas en un centro hospitalario (23). Además, hay que realizar estudios complementarios de la coagulación y de los niveles de los factores y niveles de hematíes. Hay que comprobar si el paciente tiene inhibidores del factor VIII, para determinar si el tratamiento de reposición será admitido.
- Desde hace algunos años, se está administrando desmopresina (DDAVP), en perfusión intravenosa continua de una solución isotónica de 50 ml, durante 15 min. Como medida profiláctica para la cirugía dental ofrece buenos resultados, reduciendo las complicaciones postoperatorias, por lo que se recomienda la utilización de este fármaco para la prevención del sangrado en cirugía menor (1, 24). Zanon y cols, en un trabajo, realizaron extracciones en pacientes hemofílicos y pacientes control. A todos los pacientes con hemofilia les administraron 20 mg/kg de ácido tranexámico y una infusión simple de factor VIII o IX, dependiendo del caso, para llegar a un nivel del 30% del factor ausente, antes de la extracción dental. Las complicaciones hemorrágicas no se diferenciaron de la población normal, con lo que concluyeron que se puede realizar extracciones con un menor disconfort para los pacientes (25). La aplicación de enjuagues de ácido tranexámico, son de gran utilidad, ya que estos inhiben la fibrinólisis, (se recomienda dosis de 10 mi por 2 minutos, 4 veces al día, durante una semana) (17). Entre otros agentes tópicos que también están disponibles para el control del sangrado, se encuentran la esponja de gelatina, el óxido de celulosa, los tapones de fibrina o trombina y el colágeno. No se deben prescribir analgésicos que contengan aspirina (26).

- Es importante tener en cuenta que los pacientes hemofílicos constantemente están sometidos a tratamientos de reposición o transfusiones, con lo cual tienen mayor riesgo de ser contagiados por el virus de la hepatitis o el sida, por lo que en la consulta se deben mantener las medidas de profilaxis para evitar el contagio tanto de personal como de otros pacientes que asistan a la consulta (1).
- Es recomendable realizar el máximo de tratamiento durante la terapia de reposición del factor deficitario (1). Recientemente se ha descrito un protocolo de actuación para el manejo de los pacientes con hemofilia A (tabla V) (27).
ENFERMEDAD DE VON WILLEBRAND
Es un trastorno hereditario, autosómico dominante, de una parte del complejo del factor VIII von W, necesario para la función plaquetaria normal. Como consecuencia de este déficit, la enfermedad se manifiesta como una alteración funcional plaquetaria. Es un desorden común de la sangre, por deficiencia o disfunción del factor von WiIlebrand (vWF), como consecuencia de mutaciones en el código genético de este multímero de glicoproteína. El factor vWF participa en dos funciones principales: la hemostasia primaria y la coagulación intrínseca, ya que media la adhesión de las plaquetas al sitio del daño vascular y estabiliza el factor VIII en la circulación (17). La alteración de estas funciones es expresada por bajos niveles de actividad coagulante del factor VIII en plasma (VIIIc), y por un tiempo de sangrado prolongado (28, 29). La enfermedad vW es fenotípicamente heterogénea y se clasifica en 3 tipos diferentes: Tipo I, deficiencias cuantitativas parciales; tipo II, deficiencias cualitativas; y tipo III, deficiencias cuantitativas totales (2, 6).
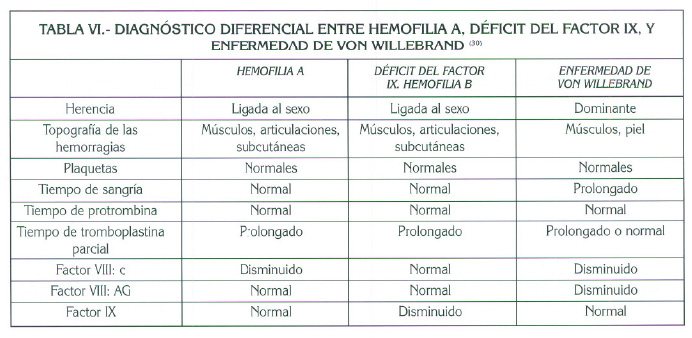
Las diferencias entre los dos tipos de hemofilia y la enfermedad de von WiIlebrand se explican en la tabla VI (30).
Manifestaciones clínicas
A diferencia de la hemofilia, las manifestaciones hemorrágicas más importantes son las mucocutáneas, mientras que las hemartrosis o hemorragias musculares sólo se observan en el tipo III. Las epistaxis, gingivorragias y metrorragias son las complicaciones más frecuentes. Puede sospecharse el diagnóstico en pacientes con antecedentes de hemorragias en mucosas e historia familiar positiva que afecta a ambos sexos. El tiempo de hemorragia es un parámetro fundamental para establecer el diagnóstico, ya que está alargado en todos los casos. El TTPA puede ser normal o alargado, dependiendo de la cantidad de factor vWF circulante (6).
Manifestaciones orales
Las manifestaciones más comunes son las hemorragias orales espontáneas y las hemorragias prolongadas tras extracciones dentales u otras intervenciones quirúrgicas bucales.
Tratamiento
El objetivo es el control de la hemorragia, y para ello se intentarán normalizar dos parámetros: el tiempo de hemorragia y la concentración de factor VIII. En el tipo I se recomienda el uso de DDAVP, mientras que en el tipo III esto no es suficiente y se utiliza crioprecipitado, que es rico en vWF, asociado a transfusión de plaquetas (6).
Manejo odontológico
El tratamiento debe ser individualizado, teniendo en cuenta el grado de las hemorragias y la gravedad de la enfermedad en cada paciente. La anestesia troncular se debe evitar por la posible formación de hematomas. En pacientes con enfermedad leve sometidos a tratamiento dental, el uso de ácido ε-aminocaproico (EACA), junto con crioprecipitado, puede reducir la hemorragia preoperatoria. El ácido tranexámico (AMCA), es otro agente antifibrinolítico sistémico; la dosis recomendada en cirugía oral es de 15-25 mg/kg, tres veces al día (1). Otras dosis recomendadas recientemente son del EACA, 60 mg/kg cada 4 a 6 horas y del AMCA 20-25 mg/kg cada 8-12 horas; pueden ser administrados por vía oral, intravenosa o tópica (31). Al igual que en la hemofilia, se puede administrar desmopresina (1-deamino-8-D- arginina vasopresina), que es un análogo sintético de la hormona hipofisaria antidiurética vasopresina (1,32). Una sola perfusión intravenosa de DDAVP de 0,3-0,4 μg/kg, diluida en 50-100 ml de solución salina, induce un rápido aumento (30 min) del nivel plasmático del factor vWF, de 3 a 5 veces por encima del nivel basal. En general las altas concentraciones de factor VIII y vWF se mantienen en plasma por lo menos de 8-10 horas (1, 17,33,34). Se pueden repetir cada 12 o 24 horas, si es necesario. Este fármaco también está disponible en concentrados subcutáneos y de administración intranasal, a dosis de 0,3 μg/kg Y 300 μg respectivamente, lo cual puede ser de utilidad para el tratamiento en casa (33, 35). En un estudio se evaluaron altas dosis de DDAVP intranasal, administrando 1,5 mg/ml, como profilaxis en procedimientos dentales, en 333 pacientes con hemofilia y enfermedad de von Willebrand tipo I. El resultado fue catalogado como excelente o bueno en el 95% de los casos. Los resultados demuestran eficacia y seguridad al utilizar el spray intranasal con altas dosis de DDAVP para el control de episodios de sangrado, en pacientes con niveles bajos de factor VIII, von WilIebrand, o ambos (36).
USO DE ANTICOAGULANTES Y ANTIAGREGANTES ORALES
En la actualidad muchas enfermedades, destacando la enfermedad isquémica del corazón, las trombosis venosas profundas y la implantación de prótesis valvulares, entre otras alteraciones son tratadas con fármacos anticoagulantes. El nivel deseado de anticoagulación varía para los diferentes procesos (17). El manejo de estos pacientes en caso de requerir extracciones dentales ha sido modificado en los últimos años, valorando el riesgo de hemorragia postextracción contra el riesgo de potenciar problemas más graves de tromboembolismo (37, 38). Actualmente se trata de mantener los niveles de anticoagulación y tratar cualquier hemorragia postextracción con medidas locales, como el colágeno (surgicel®), fibrina adhesiva y enjuagues con ácido tranexámico (38). El tiempo de protrombina (TP) es el parámetro utilizado para monitorizar el efecto del tratamiento anticoagulante oral (TAO). Actualmente se proporciona el resultado en forma de cociente normalizado internacional (INR: International Norma-lized Ratio). El INR es el cociente entre el TP del paciente expresado en segundos y un TP denominado control (de un grupo de plasmas de pacientes con coagulación normal), elevado a un parámetro conocido como índice de sensibilidad internacional (ISI) que indica la sensibilidad de la tromboplastina empleada como reactivo. Así, la , ISI (17. 39) formula es: INR= (TPpadente/ TPcontrol)ISI (17,39) .Para un TP dentro del rango normal, el INR debe ser aproximadamente de 1. Con un INR de 2 a 3, usualmente el paciente está siendo tratado para trombosis venosa profunda, y un INR superior a 3,5 es para pacientes con prótesis valvulares cardíacas (40). Entre los fármacos más comúnmente usados en el TAO, se encuentran la heparina, que es un potente anticoagulante que inhibe el efecto de la trombina y de los factores IX, X y XII activados. La mejor forma de medir el efecto de la heparina es por el tiempo de tromboplastina parcial activado. También se encuentran los fármacos cumarÍnicos: Aldocumar® (Warfarina), Dicumarol®, Sintrom® (acenocumarol), que son antagonistas de la vitamina K. La mejor forma de medir este efecto es por el TP. Además de los anticoagulantes existen fármacos inhibidores de la agregación plaquetaria, como son los que contienen ácido acetilsalicílico (Aspirina®, Tromalyt®, Adiro®) y otros antiagregantes plaquetarios como el Disgren® (Trifusal), los cuales inhiben la ciclooxigenasa plaquetaria, bloqueando así la formación de tromboxano A2 (1,41).
El 90% de las hemorragias postextracción se deben a otras causas diferentes a la terapia anticoagulante, como son: excesivo trauma operatorio de los tejidos blandos, pocas instrucciones postoperatorias, manipulación del alvéolo o de otro sitio operatorio, como colocar la lengua o succionar (esto puede causar la fibrinólisis en el sitio de la herida); el uso inapropiado de medicamentos para la analgesia, como la aspirina u otro fármaco antiinflamatorio no esteroideo, que pueden interferir con la función plaquetaria e inducir al sangrado.
RECOMENDACIONES FUNDAMENTALES EN EL MANEJO ODONTOLÓGICO DE PACIENTES TRATADOS CON ANTICOAGULANTES
En estos pacientes es importante realizar tratamiento dental preventivo para minimizar la necesidad de intervenciones quirúrgicas. Además se debe realizar una buena historia clínica, ya que las enfermedades sistémicas pueden agravar la tendencia al sangrado. A continuación se mencionan algunas pautas que se deben seguir antes del tratamiento dental: (42, 43)
q Se debe realizar una historia clínica completa, que incluya:
- Valoración de la condición médica del paciente, determinando la necesidad de administrar profilaxis antibiótica
- Presencia de factores que puedan incrementar el riesgo de sangrado
- Experiencias de sangrado tras procedimientos de cirugía oral
- Hábitos (alcohol)
q Examen oral que determine:
- Grado de urgencia del procedimiento quirúrgico;
- Valoración del estado gingival
q Analítica preoperatoria:
- Hemograma: hematÍes, leucocitos, plaquetas
- Coagulación: tasa de protrombina, TTp, INR, tiempo de sangría
q Se recomienda conocer el INR el mismo día de la intervención. El rango terapéutico de IINR está entre 2-5; para la realización de cirugías menores debe ser menor de 3,5, y el sangrado puede ser controlado con medidas locales o tópicas, que ya han sido explicadas en apartados anteriores. Pacientes con INR superior a 3,5 y con otros factores de riesgo (coagulopatÍas, aspirina, enfermedad hepática), deben ser tratados en ámbito hospitalario. (44).
q La técnica quirúrgica debe ser lo más atraumática posible
q Se recomienda utilizar sutura reabsorbible, para disminuir el riesgo de sangrado, al no ser preciso retirar los puntos
q Después de la cirugía se puede utilizar un apósito de gasa con ácido tranexámico, durante 20 minutos. Enjuagues con ácido tranexámico 1ami durante 2 minutos cada 6 horas (7 días).
q En caso de necesidad de indicar analgésico se recomienda paracetamol y/o codeÍna
q Los procedimientos de operatoria dental pueden realizarse con un INR de hasta 3. No se recomienda realizar ningún tratamiento en la cavidad bucal con un INR superior a 3,5.
q Se debe indicar al paciente medidas terapéuticas que reduzcan el riesgo de hemorragia postoperatoria como: hielo, dieta blanda, evitar enjuagues y cuerpos extraños intraorales, evitar hábitos de succión y evitar ejercicios físicos violentos (2).
q En todos los casos se recomienda la interconsulta con el médico responsable.
DISCUSIÓN
Teniendo en cuenta todo lo expuesto anteriormente, el odontólogo debe tener conocimientos básicos, para tratar a pacientes con alteraciones de la hemostasia, ya sea primaria o secundaria. Principalmente debe realizar una correcta historia clínica, para así diferenciar si se trata de un alteración de tipo hereditaria o adquirida, y así poder realizar un correcto diagnóstico de la enfermedad y un plan de tratamiento adecuado. El odontólogo debe conocer los procedimientos simples para el control de la hemorragia, como son el uso de esponja de gelatina, la celulosa-oxidasa, el ácido tranexámico y los apósitos de colágeno, entre otros. Debemos valorar en el manejo de estos pacientes, las medidas preventivas y locales, junto con el tratamiento específico para cada trastorno de la hemostasia. Es importante que hagamos participativo al paciente en su tratamiento, explicándole que con un cuidado dental apropiado y medidas profilácticas, la intervención del odontólogo será menor, reduciendo el riesgo de posibles complicaciones hemorrágicas. Antes de realizar cualquier tratamiento es recomendable realizar una interconsulta con el hematólogo que trata al paciente.
BIBLIOGRAFÍA
1. Catalano P.Trastornos hemostáticos. En: Roce L, Kaye D, editores. Medicina Interna en Odontología. 1 ed. Barcelona: Salvat; 1992. p. 431-65. [ Links ]
2. BermudoAñino L, Gutierres JL. Manejo del paciente con trastornos hemorrágicos. En: BermudoAñino L, Palma Gómez de la casa A, editores. Tratamiento del paciente odontológico con hemopatías. Madrid: Glaxo-Smith-LilineIntigraf; 2001. p. 39-54. [ Links ]
3. Silvestre Donat FJ. Alteraciones de la hemostasia. En: Bagán Sebastián JV, Ceballos Salobreña JV, Bermejo Fenoll A, Aguirre Urízar JM, Peñarrocha Diago M, editores. Medicina Oral. 1 ed. Barcelona: Masson; 1995. p. 618-23. [ Links ]
4. Guyton AC. Hemostasia y Coagulación de la sangre. En: Guyton AC, Hall JE, editores. Tratado de Fisiología Médica. 10 ed. Madrid: McGrawHill. lnteraméricana.; 2002. p. 509-21. [ Links ]
5. Cutando A, Gil-Montoya JA. El paciente dental con alteraciones de la hemostasia. Revisión de la fisiopatología de la hemostasia para odontólogos. Med Oral 1999;4:485-93. [ Links ]
6.Castillo Cofiño R, Ordinas Bauzá A, Reverter Calatayud JC, Vicente García V,Rocha Hernando E, Martínez-Brotons F. Enfermedades de la hemostasia. En: Farreras Valentí P, Rozman C, editores. Medicina Interna. Madrid: Harcourt; 2000. p. 2001-48. [ Links ]
7. Gallardo Castillo I, Ríos Santos JV, Bullón Fernández P. Manifestaciones Orales de las alteraciones de la Hemostasia. En: Ceballos Salobreña JV, Bullón Fernández P, Gándara Rey JM, Chimenos Kustner E, Blanco Carrión A, Martínez-Sahuquillo Márquez A, et al., editores. Medicina Bucal Práctica. 1 ed. Santiago de Compostela: Asoprogaio; 2000. p. 597-610. [ Links ]
8. Jansson JH, Boman K, Brannstrom M, Nilsson TK. High concentration of thrombomodulin in plasma is associated with hemorrhage: a prospective study in patients receiving long term anticoagulant treatment. Circulation 1997;96:2938-43. [ Links ]
9. Gresele P, Momi S, Berrettini M, Nenci GG, Scwarz Hp, Semeraro M, et al. Activated protein C prevents thrombin induced thromboembolism in mice. Evidence that activated protein C reduces intravacular fibrin accumulation through the inhibition of aditional thrombin generation. J Clin invest 1998; 101: 667-76. [ Links ]
10. Joseph R. Introducción a los trastornos hematológicos. En: Roce L, Kaye D, editores. Medicina interna en odontología. Barcelona: Salvat; 1992. p. 343-8. [ Links ]
11. González R, Lozano ML, Moraleda JM. Trastornos de la hemostasia primaria. En: Moraleda JM, editor. Hematolgía. 2 ed. Madrid: Luzán 5, S.A; 1996. p. 471-85. [ Links ]
12. Martínez-Sahuquillo Márquez A. Manifestaciones Orales de las enfermedades hematológicas. En: Bascones Martínez A, editor. Tratado de odontología. Madrid: Smithkline Beecham; 1998. p. 3173-80. [ Links ]
13. García García A. Tumores benignos de la cavidad oral. En: Bascones Martínez A, editor. Tratado de odontología. Madrid: Smithkline Beecham; 1998. p. 3966. [ Links ]
14. Philip Sapp J, Evercole L, GP W Patología oral y maxilofacial contemporánea. En. Madrid: Harcourt; 1998. p. 406-15. [ Links ]
15. Sabba C, Pasculli G, Cirulli A, Gallitelli M, Virgilio G, Resta F, et al. Hereditary hemorrhagic teleangiectasia (Rendu-Osler-Weber disease). Minerva Cardioangiol 2002; 50: 221-38. [ Links ]
16. Barak S, Katz J, Kaplan I. The C02 laser in surgery of vascular tumors of the oral cavity in children. J Dent Child 1991; 58: 293-6. [ Links ]
17. Schardt-Sacco D. Update on coagulopathies. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2000; 90: 559-63. [ Links ]
18. Sonbol H, Pelargidou M, Lucas VS, Gelbier MJ, Mason C, Roberts GJ. Dental health indices and caries-related microflora in children with severe haemophilia. Haemophilia 2001;7: 468-74. [ Links ]
19. Kleinman MB. Anti-inhibitor coagulant complex for the rescue therapy of acquired inhibitors to factor VIII: case report and review of the Iiterature. Haemophilia 2002; 8: 694-7. [ Links ]
20. Goedert JJ, Eyster ME, Lederman MM, Mandalaki T, De Moerloose P, White GC, 2nd, et al. End-stage Iiver disease in persons with hemophilia and transfusion-associated infections. Blood 2002; 100: 1584-9. [ Links ]
21. Diamondstone LS, Aledort LM, Goedert JJ. Factors predictive of death among HIV-uninfected persons with haemophilia and other congenital coagulation disorders. Haemophilia 2002; 8: 660- 7. [ Links ]
22. Scully C, Cawson R. Haemorrhagic Disorders. En: Medical Problems in Dentistry. 3era ed. Great Britain: Butterworth-Heinemann; 1993. p. 82105. [ Links ]
23. Mokhtari H, Roosendaal G, Koole R, Mauser-Bunschoten EP, van den Berg HM. [Oral surgery in hemophilia patients]. Ned Tijdschr Tandheelkd 2003; 110: 74-7. [ Links ]
24. Ehl S, Severin T, Sutor AH. DDAVP (desmopressin; 1-deamino-cys-8-D-arginine-vasopressin) treatment in children with haemophilia B. Br J Haematol 2000; 111: 1260-2. [ Links ]
25. Zanon E, Martinelli F, Bacci E, Zerbinati P, Girolami A. Proposal of a standard approach to dental extraction in haemophilia patients. A casecontrol study with good results. Haemophilia 2000; 6: 533-6. [ Links ]
26. Bodner L, Weinstein J, Kleiner A. Efficacy of fibrin sealent in patients on various levels of oral anticoagulant undergoing oral surgery. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998; 86: 421-3. [ Links ]
27. Stubbs M, Lloyd J. A protocol for the dental management of von WiIlebrand's disease, haemophilia A and haemophilia B. Aust Dent J 2001; 46: 37-40. [ Links ]
28. Sadler J, Mannucci P, Berntorp E, et al. Impact, diagnosis and treatment of von WiIlebrand disease. Thromb Haemost 2000; 84: 160-74. [ Links ]
29. Mannucci PM. How I treat patients with von Willebrand disease. Blood 2001; 97: 1915-9. [ Links ]
30. Veiga P, Caballro Barrigón M. Enfermedades hemorrágicas por alteraciones de la coagulación. En: Veiga P, editor. Pediatría. 1era ed. Madrid: Egraf; 1995. p. 649-55. [ Links ]
31. Mannucci P. Hemostatic drugs. N Engl J Med 1998; 339: 245-53. [ Links ]
32. Kaufmann J, Okshe A, Wollheim C, Gunther G, Rosenthal W, Vischer U. Vasopressin-induced von WiIlebrand factor secretion form endhotelial cells involves V2 receptors and cAMP. J Clin Invest 2000; 106: 107-16. [ Links ]
33. Mannucci P, Canciani M, Rota L, Donovan B. Response of factor VIII/von WiIlebrand factor to DDAVP in healthy subjects and patients with ha emophilia A and von WiIlebrand disease. Br J Haematol1981; 47: 283-93. [ Links ]
34. Mannucci P, Ruggeri Z, Pareti F, Capitanio A. Deamino-8-D-arginine vasopressin: a new pharmacological apprach to the manegement of haemophilia and von WiIlebrand disease. Lancet 1977; 1: 869-72. [ Links ]
35. Mannucci P. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years. Blood 1997; 90: 2515-21. [ Links ]
36. Leissinger C, Becton D, Cornell C, Jr., Cox GiII J. High-dose DDAVP intranasal spray (Stimate) for the prevention and treatment of bleeding in patients with mild haemophilia A, mild or moderate type 1 von Willebrand disease and symptomatic carriers of haemophilia A. Haemophilia 2001; 7: 258-66. [ Links ]
37. DeClerck D, Vinckier F, Vermylen J. Influence of anticoagulation on blood loss following dental extractions. J Dent Res 1992; 71: 387-90. [ Links ]
38. Bailey B, Fordyce A. Complications of dental extracctions in paitents receiving warfarina anticoagulant therapy: a controlledclinical trial. Br Dent J 1983; 155: 308-10. [ Links ]
39. Hirsh J, Dalen J, Deykin D, Poller L, Bussy H. Oral anticoagulants. Mechanism of action, clinical efectiveness and optimal therapeutic range. Chest 1995; 108: 231 S-46S. [ Links ]
40. Steinberg M, Moores JI. Use of INR to asses degree of anticoagulation in patients who have dental procedures. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1995; 80: 175-7. [ Links ]
41. Little JW, Falace DA, Miller CS, Rhodus NL. Trastornos hemorrágicos. En: Tratamiento odontológico del paciente bajo tratamiento médico. 5ta ed. Madrid: Harcourt Brace de España; 1998. p. 466-94. [ Links ]
42. Martínes-Sanz J, Brescó-Salinas M, Berini-Aytés L, Gay Escoda C. Cirugía bucal y anticoagulantes orales: una propuesta de actuación. RCOE 1998; 3: 555-62. [ Links ]
43. Halfpenny W, Fraser JS, Adlam DM. Comparison of 2 hemostatic agents for the prevention of postextraction hemorrhage in patients on anticoagulants. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001; 92: 257-9. [ Links ]
44. Crispian S, Andy G. Oral surgery in patients on anticoagulant therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 94: 57-64. [ Links ]
45. González R, Lozano ML, Moraleda JM. Diagnóstico de los trastornos de la hemostasia. En: Moraleda JM, editor. Hematología. Madrid: Luzán 5, S.A; 1996. p. 459-69. [ Links ]
CORRESPONDENCIA
Dr. Eduardo Chimenos Kustner
Vía Augusta 124, 1º 3ª
08006 Barcelona

