










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAvances en Odontoestomatología
versão On-line ISSN 2340-3152versão impressa ISSN 0213-1285
Av Odontoestomatol vol.21 no.5 Madrid Set./Out. 2005
Neurofibromatosis tipo 1 (NF1) revisión y presentación de un caso clínico con manifestaciones bucofaciales
Orozco Ariza JJ*, Besson A***, Pulido Rozo M****, Ruiz Roca JA**, Linares Tovar E**, Sáez Yuguero Mª R*****
|
INTRODUCCIÓN
El neurofibroma es la neoplasia más común de los nervios periféricos. Surge de varios tipos celulares entre los que se incluyen las células de Schwann y los fibroblastos perineurales (1).
Las neurofibromatosis constituyen un grupo de trastornos hereditarios de transmisión autosómica dominante, con una prevalencia de 1 caso por cada 3.000 nacimientos (1), cuya expresividad varía y, con frecuencia, en el 50% de los casos no hay antecedentes familiares de la enfermedad, lo que representa que aparece como el resultado de una mutación espontánea (1, 2). El gen involucrado en la NF1 está ubicado en el brazo largo del cromosoma 17, exactamente en la banda q11.2; este gen secreta una proteína conocida como neurofibromina la cual tiene como función inhibir el crecimiento celular anormal. Dicho gen contiene aproximadamente 50 exones, lo que explica la gran variabilidad de penetración de la enfermedad, y la gran variabilidad de sus características clínicas puede ser explicada por las numerosas mutaciones detectadas. Por otro lado, el responsable en la NF2 se ubica en el brazo largo del cromosoma 22, aunque no se sabe aún en cual de sus bandas.
Se han descrito al menos 8 formas de esta entidad nosológica, pero la más frecuente es la neurofibromatosis tipo I (NFI) o enfermedad de Von Recklinghausen, que constituye el 80-85% de los casos (1).
En julio de 1987, el "National institut of health concensus development conference on neurofibromatosis" estableció una nueva clasificación de las neurofibromatosis mayores, que las dividió en dos tipos (3, 4): la NEUROFIBROMATOSIS TIPO I (NF1) denominada, como se citaba anteriormente, enfermedad cutánea de Von Recklinghausen o neurofibromatosis periférica, y la NEUROFIBROMATOSIS TIPO II (NF2) antes conocida como neurofibromatosis acústica bilateral o neurofibromatosis central.
En esta misma conferencia de julio de 1987, se determinaron los siguientes criterios diagnósticos de las neurofibromatosis (3, 4):
Manchas "café con leche"
Neurofibromas
Pecas axilares (signo de Crowe) y/o inguinales
Glioma del nervio óptico
Hamartomas del iris (nódulos de Lisch)
Lesión Ósea
Familiar del primer grado con NF1
Dos o más de cualquiera de estos criterios son suficientes para diagnosticar una NF1.
La NF2 es una enfermedad muy rara con una prevalencia de 1:50.000. Para diagnosticarla es suficiente con que se presente uno de estos supuestos:
Un tumor bilateral del VIII par craneal o que el paciente tenga un familiar de primer grado con NF2 asociado a un tumor unilateral del VIII par craneal o asociado a dos de los siguientes signos: neurofibromas, schwannoma, glioma o meningioma o catarata pre-senil.
MANIFESTACIONES CLÍNICAS
Estos pacientes presentan múltiples neurofibromas que pueden aparecer en cualquier lugar de la economía, aunque son más frecuentes en la piel. El aspecto clínico puede variar desde pequeñas pápulas o grandes nódulos blandos hasta grandes masas ondulosas (elefantiasis) a nivel cutáneo. La variante plexiforme de los neurofibromas se considera patognomónica de la neurofibromatosis.
Los tumores pueden estar ya presentes en el momento del nacimiento, pero con frecuencia comienzan a aparecer durante la pubertad y pueden continuar desarrollándose lentamente durante la vida adulta, pudiéndose observar durante el embarazo un crecimiento acelerado (1).
Existe una gran variabilidad de la expresión de la enfermedad, ya que mientras unos pacientes tienen unos pocos neurofibromas otros tienen cientos de ellos (1).
Una característica importante es la presencia de manchas "café con leche" en la piel. Estas manchas varían de pocos milímetros a varios centímetros. Normalmente están presentes en el momento del nacimiento o se pueden desarrollar en los primeros años de vida. La presencia de 6 o más manchas "café con leche" de más de 1.5 cm de diámetro se considera patognomónico de la enfermedad (1).
Los nódulos de Lisch, que son unas manchas pigmentadas translúcidas que se localizan en el iris, se encuentran en casi todos los individuos afectados. Otras anomalías que se pueden observar en estos individuos son tumores del sistema nervioso central, macrocefalia, retraso mental, convulsiones, estatura corta y escoliosis (1).
Además de lo anteriormente expuesto, hay una serie de signos tales como macrocefalia, hipertelorismo, implantación baja de las orejas, retraso mental leve o dificultad escolar, que frecuentemente se asocian a la neurofibromatosis pero que no se consideran criterios diagnósticos.
MANIFESTACIONES BUCOFACIALES
Las manifestaciones bucofaciales de la neurofibromatosis tipo1, se presentan entre el 4 y el 7% de los casos. Cuando afectan a los tejidos blandos aparecen en la lengua, en el suelo de la boca, en el reborde alveolar, en el paladar y en la mucosa yugal, siguiendo este orden de frecuencia (5, 6, 7).
El hallazgo más frecuente, en este caso en la lengua, es la hipertrofia de las papilas fungiformes y lo que presuntamente podría denominarse neurofibromas, aunque éstos solo aparecen intrabucalmente en el 25% de los pacientes (1).
En el suelo de la boca pueden observarse unos nódulos de base sésil o pediculada, de consistencia firme, recubiertos con mucosa de aspecto y consistencia normal, mientras que en los rebordes alveolares suelen apreciarse unos agrandamientos de consistencia firme que pueden producir desplazamiento de los órganos dentarios. En cuanto al paladar y la mucosa yugal, se manifiestan con las mismas características que los del suelo de la boca y rebordes alveolares.
Cuando afectan a los tejidos óseos los hallazgos radiológicos son básicamente de tres tipos:
1. Como una lesión central que es considerada poco frecuente (8, 9).
2. Aparecen como el resultado de una compresión extraósea, dando lugar a lesiones tipo atrofia o erosión de la cortical con desplazamiento de órganos o gérmenes dentarios.
3. Deformidades semejantes a las displasias que aparecen en otras partes del esqueleto. Esas lesiones pueden estar o no asociadas directamente con tejido neurofibromatoso.
También es típico que se observe radiológicamente un aumento de tamaño del agujero mentoniano, así como del conducto del nervio dentario inferior (1).
Linda Lee y col. (10) exponen en un estudio retrospectivo las principales características radiológicas presentes en neurofibromatosis (tabla 1).

Las lesiones esqueléticas craneofaciales en la neurofibromatosis son múltiples. Algunas de las manifestaciones más típicas son la ausencia del ala mayor del hueso esfenoides, la depresión de la fosa temporal y la depresión o abombamiento del hueso frontal.
ASPECTO HISTOLÓGICO
En los neurofibromas se observan células fusiformes con núcleos del mismo tipo dispuestas en una matriz de tejido conectivo y mastocitos dispersos en toda la lesión (11).
En los neurofibromas plexiformes, que son un subtipo histológico característico de la neurofibromatosis se observan extensas masas de tejido nervioso entrelazadas en una matriz de tejido colágeno y mastocitos dispersos en la lesión (11).
TRATAMIENTO
La neurofibromatosis no tiene un tratamiento específico (1). Habitualmente, el tratamiento está dirigido a la prevención y manejo de las complicaciones. Los neurofibromas faciales se pueden extirpar con un objetivo estético, y cuando afectan a la cavidad bucodentaria, se deberá realizar su exéresis si producen alteraciones de su función normal (1).
Cuando se trata de neurofibromas solitarios o poco numerosos, el tratamiento de elección es la cirugía. En cambio, cuando los neurofibromas son numerosos y producen deformidades, la ayuda psicológica desarrolla un papel importante para mejorar la calidad de vida de estos pacientes. Para tratar algunos de los síntomas asociados a la neurofibromatosis se propuso como tratamiento el Ketotifeno (bloqueador de mastocitos que produce su despigmentación) (12, 13). Ricardi (12) demostró en un estudio a doble ciego que el ketotifeno a dosis de 2 a 4 miligramos diarios tiene una acción beneficiosa en los síntomas asociados a los neurofibromas (prurito, dolor y debilidad muscular).
Nakayama y cols. (14) proponen el uso de interferón gamma en aquellos pacientes con enfermedad de Von Recklinghausen casos en los que los neurofibromas son intratables o inoperables.
En cuanto al pronóstico, estas lesiones pueden sufrir una degeneración sarcomatosa hacia neurofibrosarcoma o schwannoma maligno, en el 8-15% de los casos (1). La degeneración maligna suele seguir a un tratamiento crónico o a una extirpación quirúrgica incompleta (1).
PRESENTACIÓN DEL CASO CLÍNICO
Paciente varón de 10 años de edad, de raza negra que consultó al Servicio de Estomatología, Cirugía Oral y Maxilofacial del Hospital Universitario de Cartagena de India (Colombia), por presentar un nódulo en la parte anterior del suelo de la boca, y una ligera asimetría facial producida por una hipertrofia fibrosa comprometiendo el maxilar superior y el cuerpo mandibular izquierdo, de un año de evolución. Durante este tiempo, las lesiones fueron creciendo lentamente y de forma asintomática.
Como antecedentes familiares el padre, una tía y una hermana del paciente presentan una neurofibromatosis clínicamente confirmada. En cuanto a los antecedentes personales no se encontraron datos de importancia relacionados con la enfermedad actual. En el examen físico general el paciente estaba consciente, con buen estado muscular y nutricional, con crecimiento y desarrollo de acuerdo con la edad.
A nivel cutáneo, se encontraron numerosas manchas café con leche, dispersas por todo el cuerpo y neurofibromas. El resto del examen no reveló datos de importancia para la enfermedad (Figura 1 y 2).


Presentaba además una implantación ligeramente baja de los pabellones auriculares, un leve aumento del perímetro cefálico, una depresión de la fosa temporal izquierda y una asimetría facial comprometiendo el tercio medio e inferior del lado izquierdo (Figura 3).

En el examen bucodentario se observó un nódulo de 3x2 cm de diámetro, ubicado en la porción anteromedial del suelo de la boca, asintomático, recubierto por una mucosa de aspecto y color normal, de consistencia firme. A nivel del reborde alveolar mandibular izquierdo presentaba una tumoración de 4x2 cm de diámetro, localizada en el cuerpo mandibular, desde el primer molar temporal hasta la región retromolar. En el hemimaxilar izquierdo, se apreciaba un abombamiento palatino que comprometía desde primer bicúspide hasta la tuberosidad, con similares características clínicas que el nódulo del suelo de la boca (Figura 4 y 5).
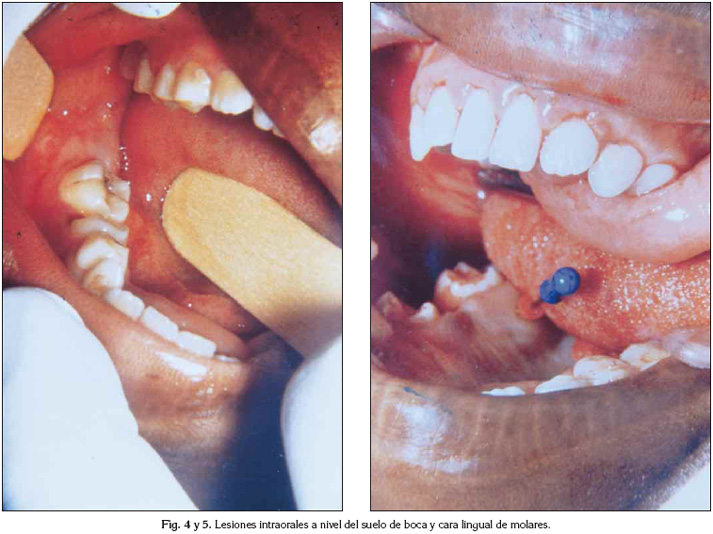
Los antecedentes familiares del paciente, las numerosas manchas café con leche, y algunos signos tales el retraso escolar, la implantación baja de las orejas y la depresión de la fosa temporal nos permitieron diagnosticar una Neurofibromatosis tipo 1 y orientar la impresión clínica de las lesiones orales hacia neurofibromas.
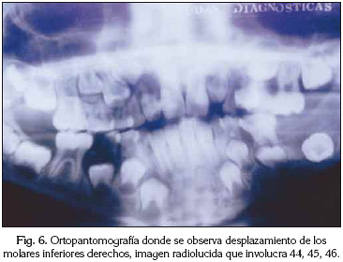
Radiológicamente se veía una imagen de compresión extraósea a nivel del cuerpo y rama ascendente mandibular izquierda, con desplazamiento de los dientes 34, 35, 36, 37 y del germen de 38. En cuanto al maxilar superior, se encontró una reabsorción ósea a nivel del órgano dentario 26 (Figura 6).
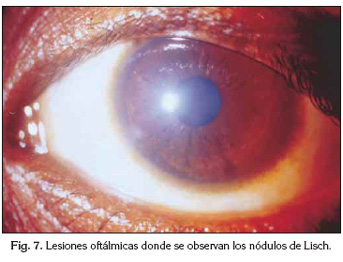
El examen oftalmológico con lámpara de hendidura reveló numerosos nódulos de Lisch lo que confirmó otro criterio diagnóstico (Figura 7).
Se llevó al paciente al quirófano para realizar, bajo anestesia general, una biopsia excisional de las lesiones tumorales así como la remodelación de los rebordes alveolares. El análisis histopatológico confirmó el diagnóstico de neurofibromas.
Con el fin de descartar la presencia de glioma del nervio óptico, se realizó una TC de órbita, sin que se revelara patología de dichos nervios.
DISCUSIÓN
Las manifestaciones de la NFI pueden ser muy diversas, tal y como se ha visto anteriormente. Así, Friedrich y cols. (15) realizaron un estudio en 48 pacientes con NFI en los que, tras examinar las radiografías panorámicas y efectuar una exhaustiva exploración intraoral, encontraron que en los 26 pacientes que tenían fibroma plexiforme había una alta prevalencia de alteraciones en la posición dentaria, así como malformaciones óseas craneomaxilofaciales. Las aberraciones numéricas y la retención de los terceros molares fue exclusivamente asociada con la afectación del nervio trigémino por el fibroma plexiforme (15).
Esas malformaciones faciales ocasionan una asimetría facial que, en un estudio realizado por Sigillo y cols. (16) en pacientes pediátricos se concluyó que se presentaba en todos los casos analizados. Además, observaron que las manifestaciones en tejidos blandos se relacionaban con el neurofibroma plexiforme mientras que si la afectación es intraósea se diagnosticaba como neurofibroma solitario (16).
Pero no solo hay una manifestación craneofacial de la NFI, sino que puede presentarse en otras partes de la economía, ocasionando una serie de complicaciones que pueden comprometer en muchos casos el pronóstico de vida del paciente (17).
El ojo es una de los órganos que más se ve afectado generalmente por gliomas que pueden progresar a pérdida de visión y a otros síntomas neurológicos. Éstos no tienen una edad de aparición determinada, aunque en un estudio de King y cols. (18) el 36% de gliomas fueron diagnosticados en niños de 6 años, por lo que recomiendan que todos los niños de 10 años de edad o jóvenes con NFI deben someterse anualmente a una revisión oftalmológica.
Y muy asociado a los tumores del nervio óptico está la pubertad precoz, que es otra de las complicaciones de la NFI. En un estudio de Virdis y cols. (19) concluyeron que la mayoría de niñas, de un total de 123, tuvieron la menarquía mucho más tarde que sus madres y que la población general.
Además de la afectación del nervio óptico, en muchas ocasiones desarrollan neoplasias en el sistema nervioso central y periférico (20).
El fibroma plexiforme suele aparecer en pacientes con NFI a nivel supraglótico, pero Osete y cols. (21), del Hospital General de Murcia, presentaron un caso con afectación laríngea. Pero no solo áreas próximas a la región facial pueden verse afectadas, pues Rizzi y cols. (22) presentaron un caso de un paciente con NFI y unos pequeños neurofibromas intestinales que le ocasionaban un dolor agudo abdominal. Y es que un tercio de los pacientes afectados por esta enfermedad presentan afectación intestinal, pero sólo el 5% debuta con sintomatología. El 10% sufren transformación maligna (22).
Weehselberger y cols. (23) presentan un caso de un varón de 17 años, con elefantiasis y nerufibromas múltiples en el hemitórax izquierdo, manifestándose así las diferentes partes corporales donde puede manifestarse la enfermedad.
CONCLUSIÓN
La neurofibromatosis fue popularizada en el año 1.971 por la novela y la película "Elephant man, de Ashley Montagu que relata la historia de Joseph Merick.
El aspecto psicológico de tales deformidades es un elemento muy importante a tener en cuenta en el difícil manejo de los pacientes con neurofibromatosis.
Es necesario llevar a cabo un seguimiento regular clínico e imagenológico para determinar la evolución de la enfermedad y prevenir las posibles complicaciones tales como ceguera por glioma del nervio óptico así como posibles malignizaciones de la lesión, como una transformación sarcomatosa (16).
La ayuda del consejo genético, el aislamiento del gen involucrado y los últimos avances de la genética molecular nos permiten esperar mucho del diagnóstico prenatal.
BIBLIOGRAFÍA
1. García García A. Tumores benignos de la cavidad oral. Bascones A (ed). En: Tratado de Odontología. Tratado de Odontología. Ed. Smithkline-Beecham. Madrid, 1999: 3962. [ Links ]
2. Lazaro C. Prenatal diagnosis of neurofibromatosis types 1. Prenat Diagn 1995; 15: 129-34. [ Links ]
3. National Institutes of Health Consensus Development Conference on Neurofibromatosis- Arch of Neurology 1988; 45: 575-8. [ Links ]
4. Roos KL, Muckway M. Neurofibromatosis. Dermatologic Clinics. Genodermatoses with Malignant Potential. 1995: 105-11. [ Links ]
5. Freedus MS, Doyle PK. Multiple Neurofibromatosis With Oral Manifestations. J Oral Surg 1975; 33: 360-3. [ Links ]
6. Vicent SD, Willisms TP. Mandibular abnormalities in neurofibromatosis. Oral Pathol 1983; 55: 253- 8. [ Links ]
7. Moore BD, Slopis JM, Jackson EF, De Winter AE, Leeds NE. Brain volume in children with neurofibromatosis type 1. Neurology 2000; 54: 914-20. [ Links ]
8. Lorson EL. Neurofibromatosis with central neurofibroma of the mandible. Review of the literature and report of case J. Oral Surg 77: 733-8. [ Links ]
9. Prescott GH, White RE. Solitary, central neurofibroma of the mandible. J Oral Surg 1970; 28: 305-9. [ Links ]
10. Linda Lee. Radigrafics features of the mandibule in neurofibromatosis: a report of 10 cases an review of the literature. Oral Surg Oral Med Oral Pathol. 1996; 81: 361-7. [ Links ]
11. Ishikawo Goro. Color Atlas of Oral Pathology – Ishiyaku Euroamerica INC. ST Louis Tokyo 1987. [ Links ]
12. Ricardi UM. Controlled multiphase trial of Ketotifen to minimize neurofibroma associated. Pain and itching. Arch Dermatol 1993; 129: 577-81. [ Links ]
13. Rause VL: Ketotifen and neurofibromatosis. Arch Dermatol 1988; 124: 651-2. [ Links ]
14. Nakayama J, Tanaka T, Arakawa F, Terao H, ron gene transfection efficiently inhibits proliferation of neurofibroma cells lines in vitro. J Dermatol 2003; 30: 181-8. [ Links ]
15. Friedrich RE, Giese M, Schmelzle R, Mautner VF, Scheuer HA. Jaw malformations plus displacement and numerical aberrations of teeth in neurofibromatosis type 1: a descriptive analysis of 48 patients based on panoramic radiographs and oral findings. Craniomaxillofac Surg 2003; 31: 1-9. [ Links ]
16. Sigillo R, Rivera H, Nikitakis NG, Sauk JJ. Neurofibromatosis type1: a clinicopathological study of the orofacial manifestations in 6 pediatric patients. Pediatr Dent 2002; 24: 575-80. [ Links ]
17. Drappier JC, Khosrotchrani K, Zeller J, Revuz J, Wolkenstein P. Medical management of neurofibromatosis 1: a cross-sectional study of 383 patients. J Am Acad Dermatol 2003; 49: 440-4. [ Links ]
18. King A, Listernick R, Charrow J, Piersall L, Gutmann DH. Optic parthway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet 2003; 122: 95-9. [ Links ]
19. Virdis R, Street ME, Bandello MA, Tripodi C, Donadio A, Vinalli AR, Cagozzi L, Garavelli L, Bernasconi S. Growth and pubertal disorders in neurofibromatosis type 1. J Pediatr Endocrinol Metab 2003; 16: 289-92. [ Links ]
20. Gold DR, Cohen BH. Brain tumors in neurofibromatosis. Curr Treat Options Neurol 2003; 5: 199- 206. [ Links ]
21. Osete Albaladejo JM, Díaz Manzano JA, Medina Banegas A, López Meseguer E, Blasco Muñoz PD. Laryngeal neurofibroma. A case report and literature review. An otorrinolaringol Ibero Am 2003; 30: 171-7. [ Links ]
22. Rizzi M, Spanghero S, Fontana A, Obersnu F, Ginanneschi U. Von Recklinhausen's disease and interstinal neurofibromatosis: a case report. Chir Ital 2003; 55: 261-5. [ Links ]
23. Wechselberger G, Bauer T, Schoeller T, Ohlbauer M, Piza-Katzer H. Elephantiasis of the thoracic wall within the scope of von Recklinghausen neurofibromatosis, case report. Wien Med Wochenschr 2003; 153: 43-5. [ Links ]
CORRESPONDENCIA
Dra. Mª Rosario Sáez Yuguero
Clínica Odontológica Universitaria "Morales Meseguer", 2ª planta.
Avda. Marqués de los Vélez, s/n. 30008 Murcia.
Teléfono 968.230.061
Fax 968.2395.65 e-mail: mrosario@um.es

