










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAvances en Odontoestomatología
versión On-line ISSN 2340-3152versión impresa ISSN 0213-1285
Av Odontoestomatol vol.21 no.6 Madrid nov./dic. 2005
Síndrome de Rendu-Osler-Weber o Telangiectasia Hemorrágica Hereditaria (HHT):
Descripción de dos casos y revisión de la literatura
Di Cosola M*, Cazzolla AP*, Scivetti M*, Testa NF*, Lo Muzio L*, Favia G** Carrillo de Albornoz A***,
Bascones A***
|
INTRODUCCIÓN
El síndrome de Rendu-Osler-Weber, también conocido como Telangiectasia Hemorrágica Hereditaria (HHT), es un desorden vascular caracterizado la presencia de telangiectasias y malformaciones arteriovenosas (MAV) o conexiones directas que predisponen a la comunicación arteriovenosa y a la hemorragia.
Se trata de una alteración vascular displásica multisistémica de carácter autosómico dominante. En su patogénesis están implicados dos genes, HHT1 y HHT2, los cuales determinan dos formas diferentes de una misma enfermedad. La variante HHT1 se origina por mutaciones en el gen endoglina (ENG), localizado en el brazo largo del cromosoma 9 (9q33- q34.1) (1, 2), mientras que HHT2 es causado por mutaciones en el gen ALK1, localizado en el brazo largo del cromosoma 12 (12q11-q14) (3).
Se presentan dos casos de telangiectasia hemorrágica hereditaria con manifestaciones orales en la lengua y en el labio inferior, sin otras lesiones sistémicas asociadas.
PACIENTES Y MÉTODOS
Dos pacientes, un hombre y una mujer, con HHT fueron referidos a nuestro centro (Fig. 1, 2). Presentaban edades de 49 y 57 años respectivamente y se caracterizan por ser el primer caso diagnosticado de HHT en su grupo familiar. Se evaluaron los registros clínicos, parámetros diagnósticos, tratamiento y seguimiento de estos dos pacientes.
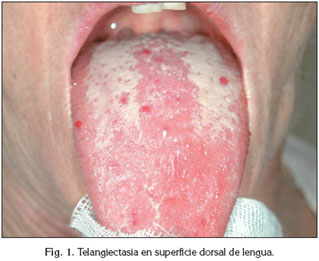
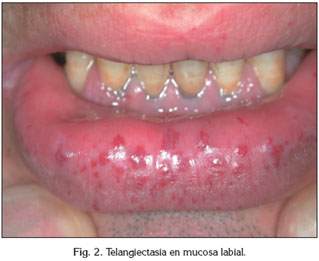
Los parámetros diagnósticos tomados en consideración para descartar patología sistémica asociada fueron: ecocardiografía de contraste para descartar comunicaciones intrapulmonares (29), tomografía computerizada, resonancia magnética del cerebro, auscultación y ecografía hepática. Ambos pacientes fueron diagnosticados de HHT1 y HHT2 por diagnóstico molecular.
El primer caso descrito concierne a la paciente de 57 años, que presentaba tras el examen oral tres raíces necróticas. El diagnóstico molecular de HHT1 había sido confirmado hacía pocos años atrás, aunque el diagnóstico clínico de sospecha le fue dado a los 19 años de edad. Las únicas manifestaciones de HHT junto con la epitaxis (15 veces por mes) aparecen en la cavidad oral, especialmente en la lengua, ya que no se pudo confirmar ninguna otra manifestación sistémica. Se trata del primer caso en su familia con este tipo de patología, excluyendo un pariente que vive en otro país.
El segundo caso descrito es el de un hombre de 49 años de edad que presentaba el tercer molar superior izquierdo impactado. Había sido diagnosticado recientemente de HHT2 por análisis molecular, aunque el diagnóstico clínico también lo obtuvo 30 años atrás. La epitaxis estaba presente con una frecuencia aproximada de 18 veces al mes, aunque la principal manifestación de la enfermedad aparecía en el labio inferior. No se encontraron otros signos de HHT confirmados tras la exploración. También era el primer caso en su familia en el cual aparecía esta patología.
Para la extracción del 28 se pautó profilaxis antibiótica con amoxicilina, con una dosis de 2 gramos/día durante 4 días previo a la exodoncia, junto con otros 6 días tras el tratamiento quirúrgico. La exodoncia se realizó con procedimientos convencionales empleando Mepivacaína como anestésico. La hemorragia se detuvo con sutura simple y compresión con gasa. Un seguimiento de tres meses fue suficiente para confirmar los resultados y la buena cicatrización de los tejidos.
DISCUSIÓN
Los genes ENG y ALK1 codifican el factor de crecimiento transformante tipo b (TGFb) expresado en las células endoteliales. La angiogénesis es regulada por ENG y ALK1 como reguladores positivos durante las fases de activación y resolución. Durante las fases de activación, las células mesenquinales se diferencian a pericitos y células del músculo liso. Se forman nuevos vasos debido a la proliferación y migración de las células endoteliales inducidas por ALK1, mientras que ALK5 induce la fase de resolución del proceso. La angiogénesis requiere un balance positivo ALK1/ALK5 y el papel de la endoglina parece ser necesario para mantener el equilibrio (4, 5). Debido a un mecanismo todavía desconocido, mutaciones genéticas en los genes ENG y ALK1 causan las alteraciones en la angiogénesis que determinan las telangiectasias y las malformaciones arteriovenosas.
Estudios in vivo en animales muestran que la expresión de ENG y ALK1 por las células endoteliales es requerida para el desarrollo de las células vasculares del músculo liso y para la comunicación entre endotelio y mesénquima (6). ALK1 es importante por su papel en la regulación de Efnb2, marcador molecular arterial. Todavía no se conoce con exactitud el mecanismo en base al cual Efnb2 y ALK1 entran en relación, aunque es precisamente esta interacción la que determina las variaciones clínicas de HHT1 y HHT2 (7). No obstante, la existencia de grupos familiares con síntomas compatibles con HHT pero sin las mutaciones genéticas, sugiere que otro gen todavía no identificado podría ser la causa de HHT en esos casos (8). Estudios recientes en ratones muestran que el incremento del factor de crecimiento de endotelio vascular (VEGF) (9) determina la formación de microvasos anormales en ENG heterocigotos (10). Sadick y cols. encontraron que VEGF se expresa no solo en al plasma, sino también en la mucosa nasal de los pacientes con HHT (11).
La base de la sintomatología clínica de la HHT es debida a la formación irregular de vasos sanguíneos. Las telangiectasias en la mucosa nasal y el sangrado nasal son los síntomas más tempranos y comunes de la HHT. El 95% de los pacientes afectados presentan epitaxis recurrente, que generalmente comienza a partir de los 12 años y se presenta con una frecuencia de 18 episodios por mes. El sangrado nasal severo puede causar anemia crónica, aunque intervalos esporádicos no requieren tratamiento. Generalmente la frecuencia y severidad del sangrado nasal incrementa con la edad, aunque algunos pacientes no refieren estos cambios. Un porcentaje similar de pacientes presenta múltiples telangiectasias en manos, cara y cavidad oral generalmente tras un período de epitaxis (12-13).
Los pacientes afectados por HHT pueden presentar telangiectasias gastrointestinales, de forma más frecuente en estómago y parte superior del duodeno; el 25% de los afectados mayores de 60 años presenta sangrado gastrointestinal generalmente asociado a melena o anemia. El sangrado es lento y persistente, y puede empeorar con la edad (14). Las trombosis o embolias son complicaciones de las malformaciones arteriovenosas y pueden aumentar con el paso del tiempo (15). Las malformaciones arteriovenosas pulmonares ocurren aproximadamente en el 30% de los individuos con HHT (16 , 17). EL 30-40% de pacientes con MAV pulmonares presentan alteraciones del sistema nervioso central, con eventos tromboembólicos tales como infarto, absceso cerebral o ataques isquémicos transitorios debido a la comunicación sanguínea. Las mujeres gestantes con MAV sin tratar presentan un riesgo mayor de hemorragia pulmonar (18). Las MAV del sistema nervioso central pueden ser congénitas.
En el 10% de los pacientes con HHT (19, 20) están presentas las MAV cerebrales, las cuales pueden presentarse a cualquier edad en forma de infartos, dolores de cabeza o hemorragia intracraneal (21, 22).
El 1% presenta MAV espinales que pueden causar hemorragia subaracnoidea, mielopatía progresiva, dolor radicular o alteraciones en esfínteres (23). Es posible encontrar comunicaciones hepáticas con una alta tasa de fracaso cardíaco, hipertensión portal, enfermedad biliar y encefalopatía portosistémica (24-27). Un estudio reciente ha identificado por tomografía computerizada anormalidades hepáticas en el 78% de pacientes con HHT (26), inclusive en casos asintomáticos.
DIAGNÓSTICO
El diagnóstico inicial de HHT continúa basándose en la presencia de signos clínicos compatibles junto con la historia familiar (28). Para el diagnóstico molecular es necesario secuenciar las regiones codificantes completas de los genes ALK1 y ENG, a pesar de que sólo es posible diagnosticar HHT en el 70% de los casos, ya que se han detectado nuevas variaciones en secuencias de desconocida significancia clínica.
El test genético no es positivo en el 100% de todos los pacientes con HHT, siendo posible encontrar mutaciones diferentes en el mismo grupo familiar (11). Estudios futuros aclararán los motivos de esta discrepancia.
TRATAMIENTO
El principal procedimiento adoptado con estos pacientes es el tratamiento sintomático del sangrado oral y nasal, junto con la prevención de las posibles complicaciones, tales como hemorragia interna, pulmonar y cerebral por MAV. El principal procedimiento para el diagnóstico de HHT es el examen clínico minucioso, valorando la presencia de telangiectasias en la cavidad oral y nasal. Para una evaluación sistémica es necesaria una ecocardiografía de contraste para descartar shunts pulmonares (29), tomografía computerizada, resonancia magnética cerebral, auscultación y ecografía hepática.
Para reducir el riesgo de fenómenos embólicos, las MAV pulmonares en las que el diámetro del vaso sea mayor de 3 mm deberán ser cateterizados, siendo necesario el seguimiento de estas lesiones debido a que pueden crecer de tamaño con el tiempo (16). Todos los procedimientos quirúrgicos para tratar las MAV pulmonares, cerebrales y hepáticas son muy peligrosos por el alto riesgo de hemorragia, defectos neurológicos o inclusive muerte que entrañan. Por estas razones, no existen protocolos quirúrgicos para tratar las lesiones de HHT, valorando en cada caso individual el tratamiento a seguir en función del riesgo. El sangrado gastrointestinal se trata con suplementos de hierro, etinilestradiol/noretindrona, danazol, ácido aminocaproico o más recientemente, por aplicación endoscópica del láser (30).
La anemia puede ser controlada de forma oral o parenteral con hierro o bien con transfusiones sanguíneas. El mejor manejo de las epitaxis leves es la aplicación diaria de lubricantes nasales, y en caso de sangrado nasal moderado es aconsejable el empleo del láser (30, 31), aunque para las formas severas puede ser necesario el uso de injertos de piel (32).
Las manifestaciones en la piel generalmente no requieren tratamiento, pero en caso de sangrado o por motivos estéticos pueden ser eliminadas con láser. Los pacientes deberán evitar los medicamentos que interfieran con la coagulación.
CONCLUSIONES
Se acepta comúnmente que las MAV pulmonares (33, 34) son más frecuentes en HHT1, y que los desórdenes hepáticos son más frecuentes en HHT2 (35-37), aunque la mayoría de las manifestaciones, como las orales, son frecuentes en ambos subtipos. Sin embargo, existen pacientes con HHT2 y MAV pulmonares, y pacientes con HHT1 y manifestaciones hepáticas, por lo que en ocasiones no se corresponde el diagnóstico genético con el comportamiento clínico (36-38).
La peculiaridad de las características de esta patología dificulta su manejo tanto por las diferentes regiones anatómicas donde se manifiesta como por el riesgo que en ocasiones entraña su terapia. Por estos motivos, es importante hacer una completa exploración de estos pacientes para plantear el tratamiento más adecuado en cada caso.
BIBLIOGRAFÍA
1. McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, et al. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat Genet 1994; 6:197-204. [ Links ]
2. Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, et al. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet 1994; 6: 205-9. [ Links ]
3. Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Helmbold EA, et al. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res 1995; 5: 21-8. [ Links ]
4. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-b type I receptors. EMBO J 2002; 21: 1743-53. [ Links ]
5. Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin receptorlike kinase (ALK)1 is an antagonistic mediator of lateral TGFb/ALK5 signaling. Mol Cell 2003; 12: 817-28. [ Links ]
6. Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, et al. Endoglin, an ancillary TGF b receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 2000; 217: 42-53. [ Links ]
7. Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptorlike kinase-1. Nat Genet 2000; 26: 328-31. [ Links ]
8. Wallace G, Shovlin C. A hereditary haemorrhagic telangiectasia family with pulmonary involvement is unlinked to the known HHT genes, endoglin and ALK-1. Thorax 2000; 55: 685-90. [ Links ]
9. Cirulli A, Liso A, DOvidio F, Mestice A, Pasculli G, Gallitelli M, et al. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol 2003 110: 29-32. [ Links ]
10. Xu B, Wu YQ, Huey M, Arthur HM, Marchuk DA, Hashimoto T, et al. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J Cereb Blood Flow Metab 2004; 24: 237-44. [ Links ]
11. McDonald J, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia. haematologica 2005; 90: 728-32 [ Links ]
12. Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary haemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32: 291-7. [ Links ]
13. Porteous M, Burn J, Proctor S. Hereditary haemorrhagic tlenagiectasia: a clinical analysis. J Med Genet 1992; 29: 527-30. [ Links ]
14. Kjeldsen A, Kjeldsen J. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterology 2000; 95: 415-8. [ Links ]
15. Vase P, Holm M, Arendrup H. Pulmonary arteriovenous fistulas in herditary hemorrhagic telangiectasia. Acta Med Scand 1985; 218: 105-9. [ Links ]
16. Haitjema T, Disch F, Overtoom T, Westermann C, Lammers J. Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med 1995; 99: 519-24. [ Links ]
17. Kjeldsen A, Oxhoj H, Andersen P, Green A, Vase P. Prevalence of pulmonary arteriovenous malformations (PAVMs) and occurrence of neurological symptoms in patients with hereditary haemorrhagic telangiectasia (HHT). J Int Med 2000; 248:255-62. [ Links ]
18. Shovlin C, Winstock A, Peters A, Jackson J, Hughes J. Medical complications of pregnancy in hereditary hemorrhagic telangiectasia. Q J Med 1995; 88: 879-87. [ Links ]
19. Haitjema T, Disch F, Overtoom T, Westermann C, Lammers J. Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med 1995; 99: 519-24. [ Links ]
20. Fulbright R, Chaloupka J, Putman C, Sze G, Merriam M, Lee G, et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. Am J Neurol Radiol 1998; 19: 477-84. [ Links ]
21. Matsubara S, Manzia J, ter Brugge K, Willinsky R, Montanera W, Faughnan M. Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. Am J Neuroradiology 2000; 21: 1016-20. [ Links ]
22. Willemse R, Mager J, Westermann C, Overtoom T, Mauser H, Wolbers J. Bleeding risk of cerebrovascular malformation in hereditary hemorrhagic telangiectasia. J Neurosurg 2000; 92: 779-84. [ Links ]
23. MontAlverne F, Musacchio M, Tolentino V, Belzile F, Riquelme C, Tournade A. Giant spinal perimedullary fistula in hereditary haemorrhagic telangiectasia: diagnosis, endovascular treatment and review of the literature. Neuroradiology 2003; 45:830-6. [ Links ]
24. Garcia-Tsao G, Korzenik J, Young L, Henderson K, Jain D, Byrd B, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343: 931-6. [ Links ]
25. Memeo M, Stabile Ianora AA, Scardapane A, Buonamico P, Sabba C, Angelelli G. Hepatic involvement in hereditary hemorrhagic telangiectasia. Abdom Imaging 2004; 29: 211-20. [ Links ]
26. Stabile Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G. Hereditary hemorrhagic telangiectasia: multidetector row helical T assessment. Radiology 2004; 23: 250-9. [ Links ]
27. Saluja S, White RI. Hereditary hemorrhagic telangiectasia of the liver; hyperperfusion with relative ischemia: poverty amidst plenty? Radiology 2004; 230: 25-7. [ Links ]
28. Shovlin C, Guttmacher A, Buscarini E, Faughnan M, Hyland R, Westermann C, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber Syndrome). Am J Med Genet 2000; 91: 66-7. [ Links ]
29. Nanthakumar K, Graham A, Robinson T, Grande P, Pugash R, Clarke J, et al. Contrast echocardiography for detection of pulmonary arteriovenous malformations. Am Heart J 2001; 141: 243-6. [ Links ]
30. Longacre A, Gross C, Gallitelli M, Henderson K, White R, Proctor D. Diagnosis and management of gastrointestinal bleeding in patient with hereditary hemorrhagic telangiectasia. Am J Gastroenterology 2003 98: 59-65. [ Links ]
30. Parkin J, Dixon JA. Laser photocoagulation in hereditary hemorrhagic telangiectasia. Otolaryngol Head Neck Surg 1981; 89: 204-8. [ Links ]
31. Shapshay S, Oliver P. Treatment of hereditary hemorrhagic telangiectasia by Nd-Yag laser photocoagulation. Laryngoscope 1984; 94: 1554-6. [ Links ]
32. Saunders, W. Septal dermoplasty for control of nosebleeds caused by hereditary hemorrhagic telangiectasia or septal perforations. Trans Am Acad Ophthalmol Otolaryngol 1960; 64: 500-6. [ Links ]
33. Berg J, Guttmacher A, Marchuk D, Porteous M. Clinical heterogeneity in hereditary hemorrhagic telangiectasia: are pulmonary arteriovenous malformation more common in families linked to endoglin? J Med Genet 1996; 33: 256-7. [ Links ]
34. Heutink P, Haitjema T, Breedveld G, Janssen B, Sandkuijl L, Bontekoe C, et al. Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity. J Med Genet 1994; 31: 933-6. [ Links ]
35. Buscarini E, Buscarini L, Danesino C, Pinatanida M, Civardi G, Quaretti P, et al. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia: Doppler sonographic screening in a large family. J Hepato 1997;26: 111-8. [ Links ]
36. McDonald J, Miller F, Hallam S, Nelson L, Marchuk D, Ward K. Clinical manifestations in a large hereditary hemorrhagic telangiectasia type 2 kindred. Am J Med Genet 2000;93:320-7. [ Links ]
37. Olivieri C, Mira E, Delu G, Pagella F, Zambelli A, Malvezzi L, et al. Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J Med Genet 2002; 39: E39. [ Links ]
38. Abdalla S, Geisthoff U, Bonneau D, Plauchu H, McDonald J, Kennedy S, et al. Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. J Med Genet 2003;40:494-502. [ Links ]

