










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAvances en Odontoestomatología
On-line version ISSN 2340-3152Print version ISSN 0213-1285
Av Odontoestomatol vol.24 n.1 Madrid Jan./Feb. 2008
Marcadores de senescencia celular en cáncer y precáncer oral
Cellular senescence markers in oral cancer and precancer
Campo-Trapero J.*, Cano-Sánchez J.**, López-Durán M.***, Palacios-Sánchez B.****, Sánchez-Gutierrez J.J.*****, Bascones-Martínez A.******
* Profesor Contratado Doctor. Facultad de Odontología. UCM.
** Profesor Asociado. Facultad de Odontología. UCM.
*** Licenciada en Odontología. UCM.
**** Profesora Colaboradora Honorífica. Facultad de Odontología. UCM.
***** Médico Adjunto del Hospital Clínico San Carlos. Madrid.
****** Catedrático. Facultad de Odontología. UCM.
Dirección para correspondencia
RESUMEN
Estudios recientes han demostrado la capacidad del organismo humano para detener el crecimiento de potenciales células cancerígenas y paralizarlas. Este mecanismo antitumoral, que actúa como freno del proceso maligno, se conocía ya en estudios de laboratorio "in vitro", pero se ha comprobado también su presencia en modelos "in vivo", tanto en ratones como en muestras de tejido de pacientes con cáncer. A este mecanismo se le denomina senescencia celular y se define como un sistema de defensa de emergencia de las células que están en camino de convertirse en cancerosas, una respuesta ante el estímulo de un oncogén. Se trataría pues de un freno a la progresión de las lesiones cancerizables, condenando a esas células a una "cadena perpetua celular". Este artículo de revisión se propone describir este mecanismo y poner al día la evidencia al respecto de este proceso, así como los marcadores de senescencia existentes en relación con el cáncer y precáncer oral.
Palabras clave: Células senescentes, detención ciclo celular, senescencia celular.
ABSTRACT
Recent studies have demonstrated the capacity of the human organism to prevent the growth of potentially carcinogenic cells, paralyzing them. This antitumor mechanism, which acts as a brake on the malignant process, was already known in lab studies "in vitro" but has now also been verified "in vivo" in mice and in tissue samples from cancer patients. This mechanism is known as cellular senescence and is defined as an emergency defense system for cells on the way to becoming cancerous, i.e., a response to the stimulation of an oncogene. These cells are sentenced to "life imprisonment", impeding the progression of premalignant lesions. This review aims to describe this mechanism and present an update of the evidence on this phenomenon in the setting of oral cancer and precancer.
Key words: Senescent cells, cell cycle arrest, cellular senescence.
Introducción
El cáncer de cabeza y cuello (CCC) representa el 5-10% de todos los procesos malignos. En las últimas décadas se ha observado un aumento significativo de la incidencia de cáncer oral y de la mortalidad asociada en Europa y muy especialmente en población adulta joven (1). En algunos países en desarrollo, casi la mitad de los pacientes en los departamentos de oncología padecen cáncer en la cavidad oral, en una gran parte debido a exposición a carcinógenos como el tabaco (2).
La mayoría de los tumores malignos de la cavidad bucal son carcinomas orales de células escamosas (COCE). Anualmente se diagnostican más de 300.000 nuevos casos de COCE a nivel mundial. Esta agresiva neoplasia epitelial esta asociada a una severa morbilidad y menos del 50% de supervivencia a largo plazo a pesar de los avances en el tratamiento quirúrgico, con radioterapia y/o quimioterapia. La posibilidad de intervenir antes de llegar a estadios avanzados podría mejorar los resultados del tratamiento (2). Estos procesos malignos van en ocasiones precedidos de lesiones cancerizables especialmente la leucoplasia. Hasta un 10% de los pacientes con leucoplasia tienen un carcinoma invasivo en la lesión (3). La prevención de la malignización de las leucoplasias se hace indispensable teniendo en cuenta la baja supervivencia del cáncer oral sobre leucoplasias (sólo del 30-40% de los pacientes permanecen vivos a los 5 años después del diagnóstico) (4).
A pesar de que la leucoplasia oral se considera desde hace décadas como una lesión cancerizable, todavía existen problemas metodológicos en identificar cuales son potencialmente malignas, debido a la ausencia de marcadores pronósticos y clínicos precisos. Aunque ciertas características clínicas e histológicas se han asociado con cierta progresión de las lesiones leucoplásicas orales, no hay una clara correlación entre los diferentes grados de displasia en relación al pronóstico, de tal manera que grados leve de displasia han evolucionado a carcinomas in situ, mientras que displasias severas no han evolucionado a carcinoma (2,5).
Debido a la variabilidad de estos marcadores histopatológicos y su falta de correlación con la clínica, se necesitan nuevos marcadores moleculares diagnósticos y pronósticos. Así, los marcadores genéticos (arrays de DNA) se perfilan en un futuro como los marcadores pronósticos ideales desde un punto de vista clínico. Sin embargo, parece que el establecimiento de marcadores moleculares mediante inmunohistoquímica podría simplificar los diagnósticos rutinarios a corto y medio plazo.
Esta aceptado ampliamente que la carcinogénesis oral es un proceso molecular muy complejo, con múltiples pasos en los que se va acumulando el daño genético lo que conlleva una alteración celular y de las señales citoplasmáticas que afectan el ciclo celular y la reparación del ADN (6). Los organismos multicelulares han desarrollado al menos dos mecanismos celulares para impedir la proliferación de células con riesgo de padecer transformaciones oncogénicas: la apoptosis o muerte celular programada y la senescencia celular. Ambos mecanismos poseen características en común, sin embargo, mientras la apoptosis mata y elimina a las células potencialmente cancerígenas, la senescencia detiene irreversiblemente su crecimiento, constituyendo barreras que las células deben vencer para progresar hacia la malignidad. Las células normales, particularmente las humanas, no sufren con frecuencia apoptosis en respuesta a daños moderados al ADN, sino que responden adoptando un fenotipo senescente (7).
De hecho se ha observado que la adquisición del fenotipo inmortal no se correlaciona con el estado histopatológico (es decir el grado de displasia), aunque si se ha observado este fenotipo en los estados más tempranos de la carcinogénesis oral (8).
En esta revisión de la literatura nos proponemos abordar el concepto de senescencia celular en el cáncer y precáncer y sus posibles conexiones con el COCE y lesiones cancerizables de la cavidad oral, así como de los marcadores de senescencia más estudiados hasta el momento.
Concepto de senescencia celular
El estado de senescencia celular se alcanza después de que las células hayan proliferado en exceso o bajo estímulos anormalmente potentes, y consiste en una pérdida irreversible de la capacidad de dividirse. Este fenómeno ha intrigado a los investigadores desde hace ya 40 años, cuando fue descrito por primera vez por Moorhead y Hayflick (9). Así fue observada por primera vez en fibroblastos obtenidos de piel y pulmón en cultivos in vitro, donde se pudo comprobar que las células se dividían, pero según envejecía el cultivo, las células dejaban de dividirse. Además de una pérdida en la capacidad de división se producía una alteración en la morfología de estas células. Establecieron que los cultivos dejaban de dividirse después de una media de 50 divisiones, dato éste conocido como limite Hayflick o fenómeno de Fase III.
En la actualidad el concepto de senescencia es aplicado en general a una detención irreversible y permanente de la proliferación y replicación celular causado por varias situaciones de stress, incluyendo el daño celular oxidativo, la alteración o acortamiento de los telómeros, daños en el ADN y fármacos quimioterápicos (como la doxorubicina o el cisplatino, que afectan a la estructura del ADN; o el taxol o vincristina cuyo diana son los microtúbulos) (10, 11).
Varias evidencias sugieren pues que la respuesta senescente evolucionó para suprimir la tumorogénesis, actuando como mecanismo de seguridad para prevenir la proliferación de células con riesgo de sufrir transformaciones neoplásicas (12). Los telómeros en mamíferos, al igual que en todos los vertebrados, están constituidos por la secuencia 5'-TTAGGG-3', repetida cientos y miles de veces al final de cada cromosoma. Tanto ellos como sus proteínas especializadas asociadas (TRF1, TRF2, POT1, TIN2, hRAP1) son esenciales para la integridad de los cromosomas.
Por mecanismos bioquímicos propios de la replicación del ADN, de 50 a 200 pares de bases del ADN telomérico dejan de ser replicados durante cada fase S. Debido a esto y a que la enzima responsable de la síntesis de novo del ADN telomérico no se expresa en la mayoría de las células humanas, los telómeros se acortan en cada ciclo celular de forma fisiológica (13). Sólidas evidencias demuestran que las células expresan el fenotipo senescente cuando uno o más de sus telómeros alcanzan una longitud crítica. Así por ejemplo, en células humanas donde la longitud del fragmento telomérico de restricción terminal promedia de 15 a 200 000 pares de bases, la senescencia celular replicativa se produce cuando dicho fragmento adquiere una longitud promedio de 5 a 7 000 pares de bases, pudiendo comprometer la integridad del cromosoma (14-16). La velocidad de acortamiento de los telómeros en sí misma estaría fuertemente influenciada por el stress celular oxidativo (17), y está especialmente relacionada con procesos de envejecimiento, siendo hoy en día muy estudiadas sus posibles relaciones con enfermedades neurodegenerativas con el Alzheimer o el Parkinson (18).
En algunas ocasiones las células pueden expresar un fenotipo senescente independientemente de contar con telómeros disfuncionales, lo que se ha denominado como "SIPS" (por Stress-Induced Premature Senescence), "senescencia prematura" o "senescencia acelerada" (17), (tras la activación de los dos principales supresores tumorales p53 y p16INK4a), como sucede en células epiteliales normales (18,19).
Las células tumorales están expuestas a diversas situaciones de estrés, por lo tanto la inducción de senescencia podría constituir un importante bloqueo a la progresión tumoral. Una fuente especial de stress se deriva de señales proliferativas aberrantes de oncogenes que podrían activar la senescencia a partir de un proceso conocido como senescencia inducida por oncogenes "OIS" (OIS por Oncogene-induced senescence). Esta respuesta celular puede ser clave para la protección celular contra el cáncer. Sin embargo su investigación se ha limitado hasta ahora a células en cultivo e in vivo con ratones que se han manipulado para que sobreexpresen un oncogén (p. ej: ras) (20).
Numerosos estudios recientes establecen que OIS sucede durante los estudios más tempranos del desarrollo tumoral tanto en modelos animales como humanos (10,21). Estas observaciones indican claramente que OIS anula el crecimiento de células estresadas oncogénicamente, manteniendo por tanto al tumor en un estado premaligno, no agresivo. Por el contrario, la ausencia de OIS, que es motivada por la mutación de las vías inductoras de senescencia, deja libre el camino a la progresión maligna mediada por oncogenes (10,20,21). La asociación de senescencia con lesiones premalignas (caracterizadas por una morfología celular normal y ausencia de crecimiento invasivo) convierten a la detección de la senescencia como un atractivo biomarcador que podría servir como un indicador pronóstico. También se debe saber que la aparición de senescencia en lesiones premalignas no es incompatible con el crecimiento del tumor, ya que esto depende del equilibrio entre proliferación y apoptosis o senescencia (10).
Otro aspecto que se desconoce todavía es cuanta actividad oncogénica es necesaria no sólo para provocar o inducir el fenotipo senescente (OIS) sino también para provocar la transformación maligna de una lesión premaligna o de tejido normal y la proporción de estos niveles entre sí. Collado y Serrano proponen que el estrés oncogénico se incrementa progresivamente durante el desarrollo tumoral y que la senescencia es activada en un punto cuando lo los tumores ya han sido iniciados, pero no han alcanzado el fenotipo maligno (10).
Vías de senescencia celular
Aunque, como se ha descrito previamente, existen diversos estímulos que pueden inducir una respuesta senescente, todos ellos parecen converger en una o ambas vías que establecen y mantienen la detención del crecimiento senescente (18) (22):
Vía de ARF-p53P53
p53 es un mediador crucial de las respuestas celulares a daños en el ADN, incluyendo también la respuesta senescente, ya que la pérdida de la función p53 retrasa la senescencia replicativa de células humanas. Los telómeros disfuncionales también activan muchos componentes de la respuesta mediada por p53 y la respuesta senescente a los telómeros disfuncionales requiere de la integridad de esta vía. Las ROS (Reactive Oxygen Species) incluyen iones de oxígeno y radicales libres, provocando efectos mutagénicos en la vía RAS y también inducen la senescencia celular (23).
Así RAS oncogénico sobreexpresado puede provocar una respuesta dependiente de p53, en este caso por la producción de altos niveles de ADN dañado por ROS. Sin embargo RAS oncogénico también puede inducir p16INK4a, que es un activador de la vía pRB, lo que proporciona una segunda barrera a la proliferación de células potencialmente oncogénicas. Por lo tanto, al menos en algunas células, la inducción de senescencia por daño en el ADN, disfunción de los telómeros y posiblemente sobreexpresión de oncogenes convergen en la vía p53, que es necesaria y suficiente para establecer y mantener el arresto senescente (Figura 1). Aunque este estado senescente no puede ser revertido por señales fisiológicas, en esas células puede ser revertida por la pérdida de la función de p53 (12,18).

Vía de p16INK4a-pRB
Aunque la inactivación de p53 revierte la detención en la proliferación celular en algunas células, fracasa en otras y esto es porque se ha visto que esas células expresan p16INK4a, que es un inhibidor del ciclo celular y un regulador positivo de pRB. p16INK4a es inducido por una gran variedad de estímulos estresantes, incluido la sobreexpresión de oncogenes como RAS. Los cultivos celulares indican que p16INK4a impide la reversibilidad de la senescencia por inactivación de p53. Así pues este supresor de tumores (p16INK4a) y presumiblemente la vía pRB que activa, proporciona una formidable barrera a la proliferación celular, que no puede ser derrotada por la pérdida de la función de p53 (18).
Existen células que permanecen en estado senescente a pesar de la inactivación de p53. Esto es así porque esas células han entrado en senescencia a través de la vía p16INK4a-pRb que es una vía irreversible para la célula aunque exista inactivación de p53, pRb o ambas (18). En el proceso senescente, los complejos pRb-E2F, o lo que es lo mismo la pRb activa, van a producir el desarrollo de focos densos de heterocromatina (SAHFs por Senescence –associated heterochromatin foci) que produce la represión de varios genes implicados en el avance del ciclo celular (Figura 2). Además, un vez formados esos focos de heterocromatina parece que no necesitan la activación de p16INK4a o pRb para seguir en esa disposición lo que mantiene de manera permanente el estado senescente (18).

Marcadores de senescencia celular en cáncer y precáncer oral
Se han observado marcadores moleculares con una expresión progresivamente aumentada según se progresa en la carcinogénesis oral (p. ej. p53) pero sin encontrarse un marcador que aparezca exclusivamente en una lesión cancerizable pero no en la lesión maligna. Warnakulasuriya y cols, realizaron un metaanálisis de siete estudios encontrando sobreexpresión de p53 en un 47% de las lesiones cancerizables orales, porcentaje este mucho mayor que el de malignización de lesiones cancerizables como la leucoplasia (24). En este sentido, hay estudios que han demostrado la inexistencia de la proteína p53 (mutada) por medios inmunohistoquímicos en lesiones malignas de cabeza y cuello.
También se han presentado discordancias en cuanto a la eficacia de p21 como marcador pronóstico ya que en algunos estudios se ha observado su expresión como favorable para el pronóstico de CCC mientras que otros estudios asocian esta sobreexpresión con un fenotipo tumoral más maligno (25). Por otro lado hay estudios que han presentado como marcadores de progresión el aumento expresión de EGFR y de TGF-a, pero con unos resultados y rangos de porcentaje muy poco precisos para determinar en que situación de malignidad nos encontramos (26).
Se han observado células senescentes en lesiones premalignas de pulmón, páncreas y piel, y sin embargo no se han observado o lo hacen muy débilmente en carcinomas de tales tejidos. Parece pues que cuando una célula pierde su respuesta senescente pasa de un estado benigno (lesión premaligna) a un estadio de malignidad (carcinoma), aunque la complejidad del proceso carcinogénico no parece indicar que esto se produzca como un proceso causa-efecto sino como una expresión más de la carcinogénesis (20). Kang y Park establecieron un modelo de carcinogénesis oral in vitro para describir los mecanismos moleculares por los que los factores ambientales facilitan la aparición del cáncer oral. Así estimularon a queratinocitos humanos normales (NHOK por normal human oral keratinocytes) mediante virus de papiloma (VPH) de alto riesgo y carcinógenos químicos. Tras la introducción del genoma del VPH, las células puentean el punto de control de senescencia y entraban en un ciclo vital prolongado, pero no inmortal, durante el cual los telómeros continuaban acortándose. En unos pocos clones inmortales encontraron una elevación marcada de la actividad de telomerasa (hTERT por human telomerase reverse transcriptase) y estabilización de los telómeros. Además las oncoproteínas E6 y E7 del VPH de "alto riesgo" alteraban el control del ciclo celular y la reparación del ADN en los HOK inmortalizados y favorecía la aparición de mutaciones a causa de la inestabilidad genómica, pero este efecto no lo conseguía por si mismo sino en combinación con carcinógenos químicos. Concluyen que la carcinogénesis oralse debe a una serie de alteraciones genéticas discretas a consecuencia de un continuado cambio genotóxico por factores de riesgo ambientales (27).
En un estudio realizado mediante análisis por inmunohistoquímica de ciclina D1, p16 y pRb en secciones de tejido en parafina de 220 COCE, 90 leucoplasias y 81 muestras de tejido oral normal se observó que la transición de lesiones cancerizables a un estadio maligno estaba asociado con el fenotipo pRb-/cyclin D1+(OR = 2.294, p = 0,001) y fenotipo p53+ (OR = 2.230, p = 0,002). La pérdida de pRb y acumulación de p53 (pRb-/p53+) están asociadas con una progresión histológica de los tumores y de la adquisición de un potencial invasivo. El análisis multivariante reveló que el fenotipo pRb-/p53+ fue el más adverso pronosticador de supervivencia libre de enfermedad (28).
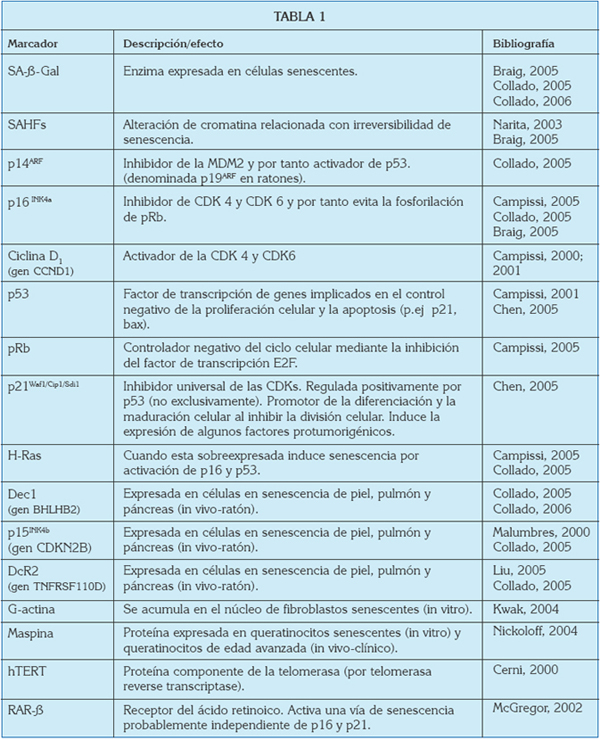
El estudio de la senescencia ha contribuido pues al establecimiento de numerosos marcadores que son útiles en la detección de esta respuesta no sólo in vitro sino también in vivo (Tabla 1) (10,18,20,29,30). A continuación comentaremos sólo aquellos marcadores de vías/fenotipos senescentes que más evidencia científica tienen en la actualidad y más posiblemente relacionados con el precáncer y cáncer oral.
ß-galactosidasa asociada a senescencia (SA-ß-gal por Senescence Associated ß-galactosidase)
El ensayo más ampliamente utilizado para senescencia es la detección histoquímica de la actividad de la ß-galactosidasa a pH 6.0, llamada SA-ß-gal. La actividad de la ß-galactosidasa se deriva del aumento del contenido lisosomal de las células senescentes, que posibilita la detección de la ß-galactosidasa lisosomal a pH subóptimo, pH 6.0 (pH 4.0 es el óptimo) (31). Se considera un marcador de senescencia tanto in vitro como in vivo (10).
Vías P16 INK4a-Rb y ARF-p53
Investigaciones de las vías de señalización que llevan a la OIS han mostrado que la vía p16-Rb (retinoblastoma) y la ARF-p53 son responsables de la detención en la proliferación que caracteriza la senescencia (18). Estas dos vías se consideran cruciales para la supresión tumoral y suelen estar mutadas en tumores. Una elevación en los niveles de p16INK4a, ARF y p53 se relaciona con OIS en tejido animal así como en lesiones premalignas humanas. Pero al estar en la parte más alta de la cascada (ver figura 2), hay situaciones en las que células tumorales podrían haber adquirido mutaciones en zonas inferiores de la cascada que bloqueen la senescencia a pesar de niveles elevados de p16 INK4a y ARF-p53 (32). Así en mucosa oral displásica se ha observado niveles de expresión elevada de p16 INK4a y en los carcinomas in situ (estudio clínico) pero no en los COCE (19).
También se ha observado experimentalmente que la expresión de la proteína p53 mutada aparece elevada en los tejidos epiteliales displásicos indicando un evento temprano en la carcinogénesis oral (33). Un estudio sobre CCC, observó que la prevalencia de mutaciones de p53 previas a la invasión no se incrementaba según el estadio clínico del tumor y que por lo tanto dichas alteraciones carecían de significado pronóstico (34).
Por todo ello es muy importante encontrar marcadores, que al contrario de p16INK4a y ARF-p53, no estén implicados en el inicio de la senescencia sino en puntos finales de estas vías de senescencia (10).
Focos de heterocromatina asociados a Senescencia (SAHFs por Senescente-associated heterochromatin foci)
Recientemente cambios epigenéticos asociados con una alteración global de la heterocromatina durante la senescencia se han relacionado con la irreversibilidad que caracteriza la respuesta senescente (20,35). Esta alteración está iniciada por RB y resulta en una estable y permanente represión de genes cruciales en la proliferación como son los factores de transcripción de la familia E2F. Estas alteraciones genómicas pueden ser visualizadas microscópicamente y se conoce con el término de focos de heterocromatina asociada a senescencia (SAHFs). Constituye un aspecto morfológico de los núcleos de las células senescentes que se han empleado exitosamente para identificar la aparición de IOS "in vivo" (20,21).
Maspina
Se ha demostrado que interacciona con el colágeno tipo I y tipo III, lo que se relaciona con su acción antiangiogénica en la matriz extracelular (36). La expresión de maspina no parece ser universal sino que aparece en determinados tipos celulares: células mioepiteliales en mama, células basales de próstata, epitelio glandular y de mucosa en tracto gastrointestinal y capas epidérmicas de la piel (30). La expresión de esta proteína se ha observado también incrementada de manera gradual en muestras de piel normal de pacientes a medida que se incrementaba la edad (30). Su expresión, sin embargo no se ha visto expresada en fibroblastos senescentes, indicando que podría ser un marcador principalmente asociado al tejido epitelial.
Telomerasa
Es una ribonucleoproteína con dos componentes con actividad enzimática a nivel nuclear. La telomerasa ARN (TR, telomerase RNA) y una proteína transcriptasa inversa de la telomerasa (hTERT, human telomerase reverse transcriptase protein). La telomerasa añade repeticiones hexaméricas de ADN (TTAGGG) a las terminaciones de los telómeros lo que compensa la pérdida progresiva de las secuencias teloméricas inherentes a la replicación del ADN (37). La actividad de la telomerasa ha sido detectada en una variedad de tejidos somáticos, incluyendo células hematopoyéticas, queratinocitos basales, y células basales en endometrio, esófago, próstata y esófago (38). El inicio de senescencia se acompaña de la inducción de p16INK4a y esto sucede en varios sistemas de cultivo diferentes empleados. Los resultados del estudio de Kang y cols indican que la SR de los NHOK está asociada con la pérdida de la actividad de la telomerasa seguida de un acortamiento limitado de los telómeros. (39).
RAR-ß
Corresponde a los receptores del ácido retinoico (RAR-ß, Por Retinoic Acid Receptor-ß) ya que los retinoides inducen la sobrerregulación de algunos genes que inducen senescencia en células de cáncer de mama sin producir efectos secundarios (como al contrario produce la senescencia inducida por p21) (40). RAR-ß se ha visto dependiente del ácido retinoico en mucosa oral normal, se observa constitutivamente expresado en displasias mortales, y completamente suprimida en displasias inmortales y COCE (8). Esto señala a la expresión de RAR-ß como un factor importante para mantener el fenotipo mortal. No está claro como están implicados los receptores de los retinoides en la senescencia y cuales son sus genes diana. Parece improbable que p16 INK4a sea un gen diana de RAR-ß porque p16 INK4a está expresada en mucosa oral normal en ausencia de RAR-ß (8). RAR-ß incrementa su expresión a medida que la célula se va acercando a los finales estadios de entrada en el estado senescente, mientras que p16 es altamente expresado incluso en los estados de mucosa normal, considerándosele una función importante para el inicio de la senescencia (8).
G-actina
También se ha observado in vitro (cultivos de fibroblastos humanos senescentes) la expresión nuclear de G-actina como marcador de senescencia incluso de manera más precoz que el incremento endógeno de la enzima ß-galactosidasa (SA-ß-gal) (29).
Mediante el empleo de microarrays de ADN se han podido seleccionar marcadores "in vivo" que mostraban cambios en la expresión génica que solamente tenían lugar durante la senescencia pero no cuando esta senescencia estaba inhibida. Estos marcadores se probaron en modelos animales de tumorogénesis provocadas por activación oncogénica endógena de HRAS o KRAS. Entre ellos destacan p15 INK4b, DCR2 y DEC-1, que mostraron niveles de expresión aumentados de forma específica en lesiones premalignas de piel, pulmón y páncreas pero no o de forma muy débil en tumores (20).
p15 INK4b
Es un inhibidor de las CDK (por cyclin dependent Kinase) que se correlaciona con la presencia de otros marcadores de senescencia especialmente SA-ß-galactosidasa y SAHFs. También se ha descrito que p15INK4b esta implicado en la detención del crecimiento celular que se impone durante la senescencia inducida por Ras y de oponerse a la transformación por Ras al menos en cultivos celulares (41).
DCR2
Es un gen inducido por p53 y es uno de los receptores señuelo del ligando- inducido por apoptosis relacionado con el factor de necrosis tumoral (TRAIL por tumour necrosis factor-related apoptosis-inducing ligand). DCR2 suprime la apoptosis inducida por TRAIL y parece regular la quimiosensiblidad (42).
DEC1
Es un factor de transcripción implicado en el control de ritmos circadianos que también se ha visto implicado en diferentes vías de señalización (20). La principal ventaja de estos tres últimos marcadores, al contrario de lo que sucede con SA-ß-gal o SAHFs, es que pueden ser fácilmente detectados en tejido incluido en parafina empleando técnicas convencionales de inmunohistoquímica (10). El uso de los marcadores de OIS en la clínica podría ser útil en la detección del cáncer en estadios tempranos y la pérdida de estos marcadores sería indicativo de progresión tumoral a un estadio maligno. Collado y Serrano proponen el concepto de "Índice de senescencia". Teóricamente los tumores que tienen un "elevado índice de senescencia" tendrían pues un mejor pronóstico que aquellos con un "índice de senescencia" bajo que se asociarían con lesiones agresivas que requerirán una intervención terapéutica inmediata (10).
Los marcadores de senescencia podrían también ser empleados para monitorizar la eficacia de determinados fármacos que inducen senescencia, ya que parece ser que estos agentes quimioterápicos provocan senescencia cuando son utilizados a dosis bajas y apoptosis cuando se emplean a altas dosis (43,44).
Los avances más significativos en el entendimiento del proceso de OIS se han producido en los últimos años, pero todavía se deben realizar muchos estudios para comprender más claramente las vías implicadas en la OIS y poder obtener nuevos marcadores de senescencia. Una posibilidad muy prometedora podría ser la identificación de factores solubles liberados por las células senescentes que podrían ser cuantificados en fluidos corporales como sangre, orina o saliva (10).
A nivel del cáncer oral se abre pues una importante línea de trabajo debido a que como ya se ha mencionado no existe en la actualidad ningún marcador diagnóstico ni pronóstico fiable de las lesiones leucoplásicas que suelen preceder al COCE. El proceso multifactorial y multifásico de la carcinogénesis oral probablemente implique la alteración funcional de miembros reguladores del ciclo celular en combinación con la salida de las vías de señalización de apoptosis y senescencia celular, por lo que además de estudios in vitro con cultivos celulares deben de realizarse estudios clínicos mediante arrays de ADN como se están ya realizando en otros cánceres de la economía humana. La descripción de las alteraciones moleculares y el entendimiento de las consecuencias de la desregulación del ciclo celular en los queratinocitos orales malignos nos podrá proporcionar enfoques novedosos diagnósticos y terapéuticos.
Conclusiones
Los estudios revisados parecen indicar que la senescencia celular inducida por oncogenes (OIS) sería un mecanismo supresor de tumores activados oncogénicamente que podría interrumpir la tumorogénesis a nivel premaligno, como demuestra el hecho de que un gran número de células en lesiones cancerizables entran en senescencia, mientras que en las células de los tumores malignos no sucede esto, o sucede de forma muy débil, debido a la pérdida de efectores de vías senescentes como p16 INK4a o ARF-p53. Un mayor entendimiento de los estímulos que inducen la senescencia celular podría permitir en el futuro ser utilizados en la prevención y tratamiento del cáncer, de la misma manera a lo que ya esta sucediendo con los mecanismos apoptóticos celulares. En el caso del cáncer oral donde no existen todavía marcadores moleculares diagnósticos y pronósticos eficientes, este mecanismo podría proporcionar a corto plazo datos muy esperanzadores, por lo que son necesarios estudios clínicos que determinen cuales de los marcadores de senescencia conocidos en la actualidad pueden ser útiles en la carcinogénesis oral.
Bibliografía
1. Oliver RJ, Dearing J, Hindle I. Oral cancer in young adults: report of three cases and review of the literature. Br Dent J. 2000;188(7):362-5. [ Links ]
2. Sudbo J, Reith A. The evolution of predictive oncology and molecular-based therapy for oral cancer prevention. Int J Cancer. 2005;115(3): 339-45. [ Links ]
3. Femiano F, Scully C. DNA cytometry of oral leukoplakia and oral lichen planus. Med Oral Patol Oral Cir Bucal. 2005;10 Suppl 1:E9-14. [ Links ]
4. Scully C, Porter S. ABC of oral health. Oral cancer. BMJ. 2000;321(7253):97-100. [ Links ]
5. Aguado A, Seoane JM, Suárez JM, Balboa JL. Terapéutica quirúrgica del precáncer oral. In: Bascones A, Seoane JM, Aguado A, Suárez JM, editors. Cáncer y precáncer oral. Madrid: 2003: 289-306. [ Links ]
6. Bettendorf O, Piffko J, Bankfalvi A. Prognostic and predictive factors in oral squamous cell cancer: important tools for planning individual therapy? Oral Oncol. 2004;40(2):110-9. [ Links ]
7. Campisi J, Kim SH, Lim CS, Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol. 2001;36(10):1619-37. [ Links ]
8. McGregor F, Muntoni A, Fleming J et al. Molecular changes associated with oral dysplasia progression and acquisition of immortality: potential for its reversal by 5-azacytidine. Cancer Res. 2002;62(16):4757-66. [ Links ]
9. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961; 25:585-621. [ Links ]
10. Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6(6):472-6. [ Links ]
11. Alberts B, Johnson A, Lewis J et al. El ciclo celular y la muerte celular programada. In: Omega: Barcelona, editor. Biología molecular de la célula. 2004: 983-1026. [ Links ]
12. Campisi J. Cancer, aging and cellular senescence. In Vivo. 2000;14(1):183-8. [ Links ]
13. Kim Sh SH, Kaminker P, Campisi J. Telomeres, aging and cancer: in search of a happy ending. Oncogene. 2002;21(4):503-11. [ Links ]
14. Mathon NF, Lloyd AC. Cell senescence and cancer. Nat Rev Cancer. 2001;1(3):203-13. [ Links ]
15. Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63(11):2705-15. [ Links ]
16. Wei S, Wei S, Sedivy JM. Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts. Cancer Res. 1999;59(7):1539-43. [ Links ]
17. von Zglinicki T, Saretzki G, Ladhoff J et al. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005;126(1):111-7. [ Links ]
18. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513-22. [ Links ]
19. Natarajan E, Saeb M, Crum CP et al. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol. 2003;163(2):477-91. [ Links ]
20. Collado M, Gil J, Efeyan A et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. [ Links ] (21) Braig M, Lee S, Loddenkemper C et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660-665. [ Links ]
22. Campisi J, Warner RH. Aging in mitotic and post-mitotic cells. In: Gilchrest BA, Bohr VA, editors. The role of DNA damage and repair in cell aging. 2001: 1-16. [ Links ]
23. Lee AC, Fenster BE, Ito H et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274(12):7936-40. [ Links ]
24. Warnakulasuriya KA, Tavassoli M, Johnson NW. Relationship of p53 overexpression to other cell cycle regulatory proteins in oral squamous cell carcinoma. J Oral Pathol Med. 1998;27(8):376-81. [ Links ]
25. Reibel J. Prognosis of oral pre-malignant lesions: significance of clinical, histopathological, and molecular biological characteristics. Crit Rev Oral Biol Med. 2003;14(1):47-62. [ Links ]
26. Srinivasan M, Jewell SD. Evaluation of TGF-alpha and EGFR expression in oral leukoplakia and oral submucous fibrosis by quantitative immunohistochemistry. Oncology. 2001;61(4):284-92. [ Links ]
27. Kang MK, Park NH. Conversion of normal to malignant phenotype: telomere shortening, telomerase activation, and genomic instability during immortalization of human oral keratinocytes. Crit Rev Oral Biol Med. 2001;12(1):38-54. [ Links ]
28. Soni S, Kaur J, Kumar A et al. Alterations of rb pathway components are frequent events in patients with oral epithelial dysplasia and predict clinical outcome in patients with squamous cell carcinoma. Oncology. 2005;68(4-6):314-25. [ Links ]
29. Kwak IH, Kim HS, Choi OR et al. Nuclear accumulation of globular actin as a cellular senescence marker. Cancer Res. 2004;64(2):572-580. [ Links ]
30. Nickoloff BJ, Lingen MW, Chang BD et al. Tumor suppressor maspin is up-regulated during keratinocyte senescence, exerting a paracrine antiangiogenic activity. Cancer Res. 2004;64 (9): 2956-61. [ Links ]
31. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113 ( Pt 20):3613-22. [ Links ]
32. Masumoto N, Fujii T, Ishikawa M et al. P16 overexpression and human papillomavirus infection in small cell carcinoma of the uterine cervix. Hum Pathol. 2003;34(8):778-83. [ Links ]
33. Okazaki Y, Tanaka Y, Tonogi M, Yamane G. Investigation of environmental factors for diagnosing malignant potential in oral epithelial dysplasia. Oral Oncol. 2002;38(6):562-73. [ Links ]
34. Bosch FX, Ritter D, Enders C et al. Head and neck tumor sites differ in prevalence and spectrum of p53 alterations but these have limited prognostic value. Int J Cancer. 2004;111(4):530-8. [ Links ]
35. Narita M, Nunez S, Heard E et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703-16. [ Links ]
36. Blacque OE, Worrall DM. Evidence for a direct interaction between the tumor suppressor serpin, maspin, and types I and III collagen. J Biol Chem. 2002;277(13):10783-8. [ Links ]
37. Cerni C. Telomeres, telomerase, and myc. An update. Mutat Res. 2000;462(1):31-47. [ Links ]
38. Harle-Bachor C, Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc Natl Acad Sci U S A. 1996;93(13):6476-81. [ Links ]
39. Kang MK, Kameta A, Shin KH et al. Senescence occurs with hTERT repression and limited telomere shortening in human oral keratinocytes cultured with feeder cells. J Cell Physiol. 2004; 199(3):364-70. [ Links ]
40. Chang BD, Swift ME, Shen M et al. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci U S A. 2002;99(1):389-94. [ Links ]
41. Malumbres M, Perez DC, I, Hernandez MI et al. Cellular response to oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor p15 (INK4b). Mol Cell Biol. 2000;20(8):2915-25. [ Links ]
42. Liu X, Yue P, Khuri FR, Sun SY. Decoy receptor 2 (DcR2) is a p53 target gene and regulates chemosensitivity. Cancer Res. 2005;65(20):9169-75. [ Links ]
43. Rebbaa A, Zheng X, Chou PM, Mirkin BL. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene. 2003;22(18): 2805-11. [ Links ]
44. Zheng X, Chou PM, Mirkin BL, Rebbaa A. Senescence-initiated reversal of drug resistance: specific role of cathepsin L. Cancer Res. 2004;64 (5):1773-80. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. Julián Campo Trapero
Departamento de Medicina y Cirugía Bucofacial.
Facultad de Odontología (UCM)
Plaza Ramón y Cajal, s/n
28040 Madrid
email: jcampo@odon.ucm.es
Fecha de recepción: Diciembre 2007.
Aceptado para publicación: Diciembre 2007.

