










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.78 no.8 ago. 2003
COMUNICACIÓN CORTA
SÍNDROME DE MÚLTIPLES PUNTOS BLANCOS
EVANESCENTES DE DIFÍCIL DIAGNÓSTICO
MULTIPLE EVANESCENT WHITE DOT SYNDROME.
A DIAGNOSTIC DILEMMA
VÁZQUEZ MAROUSCHEK C1, LÓPEZ CHECA F2
| RESUMEN Objetivo/Métodos: Presentamos un caso de Síndrome de múltiples puntos blancos evanescentes (SMPBE) que no fue diagnosticado inicialmente. Se confirmó al año y medio de seguimiento, por las características clínicas, angiográficas, y electrofisiológicas. Destacamos las dificultades para llegar a un diagnóstico, especialmente porque nuestro caso presenta variaciones con respecto a la descripción inicial. Palabras clave: Síndrome de múltiples puntos blancos evanescentes, coriorretinopatía serosa central, papilitis, neurorretinitis.
| SUMMARY Purpose/Method: We present a patient with multiple evanescent white-dot syndrome (MEWDS). On presentation the patient was not initially diagnosed. The diagnosis was established eighteen months later by the clinical, angiographic and electrophysiologic features. We emphasize the difficulties to establish the diagnosis, especially in those cases that like ours present differences with regard to the initial description of the syndrome. Key words: Multiple evanescent white-dot syndrome, Central serous choroidopathy, Papillitis, Neuroretinitis.
|
Recibido: 11/11/02. Aceptado: 20/8/03.
1 Licenciado en Medicina.
2 Doctor en Medicina.
Los autores manifiestan que no tienen interés comercial ni han recibido apoyo económico en el presente trabajo.
Correspondencia:
Carmen Vázquez Marouschek
C/. Fray Francisco de Pareja, 19
41007 Sevilla
España
E-mail: Carmenvm@msn.com
INTRODUCCIÓN
El Síndrome de múltiples puntos blancos evanescentes (SMPBE) fue descrito por Jampol y cols en 1984, como causa rara de pérdida aguda de agudeza visual (AV), en jóvenes, a veces bilateral, reversible, de etiología desconocida y pronóstico favorable. La recuperación visual se produce siempre al cabo de unos meses (1).
La aparición de puntos blancos múltiples, de tamaño de 100 a 200 micras, a nivel del epitelio pigmentario o de la retina profunda, que pueden constituirse en grupos de lesiones todavía más pequeñas, que desaparecen al cabo de unas semanas fue lo que le dio el nombre (1). Tienden a localizarse alrededor de la fóvea y en la región de las arcadas vasculares, y menos manifiesto por fuera de ellas (1). La mácula suele aparecer con un patrón granular. Pueden existir infiltraciones perivasculares y células inflamatorias en el vítreo posterior (1,2).
Se produce un agrandamiento de mancha ciega y las pruebas electrofisiológicas se alteran. Posteriormente vuelven a la normalidad. La angiografía fluoresceínica (FAG) muestra hiperfluorescencia temprana con tinción tardía de las lesiones, y cada lesión parece estar formada por múltiples áreas pequeñas de hiperfluorescencia. En la mácula pueden aparecer defectos de tipo ventana. Puede aparecer pérdida de colorante de los capilares retinianos y papilares (1,2). Las lesiones desaparecen sin dejar cicatrices y normalmente la FAG retorna a la normalidad cuando se recupera la AV (1).
Existe posibilidad de aparición de neovasos subretinianos de localización macular (3) y papilar (4).
Presentamos un caso de SMPBE de difícil diagnóstico y evolución final que difiere de la descripción inicial.
CASO CLÍNICO
Mujer de 25 años con cuadro brusco de visión borrosa, en ojo derecho (OD), metamorfopsias, y alteración en la visión de los colores. Se diagnosticó, en urgencias, como inicio de una coriorretinopatia serosa central. Allí su AV OD: 2/3 y OI: 1.
A la semana, en consultas externas, se mantiene el diagnóstico, siendo su AV OD: de contar dedos a 2 metros.
Al mes de inicio, se realiza FAG y retinografías (figs. 1 y 2). La afectación de polo posterior, junto con el edema de papila, sugieren ahora una neurorretinitis y también un proceso vasculítico. La campimetría de OD muestra entonces, agrandamiento de mancha ciega y escotoma superior entre 20-30º. OI inicia una afectación mínima que posteriormente no se mantiene. Los potenciales evocados visuales están alterados concordantes con afección de mácula y nervio óptico y esto en grado mucho menor se produce también en OI.

Fig. 1. Angiografía fluoresceínica que se corresponde con la figura 2: áreas puntiformes
de hiperfluorescencia en polo posterior con difusión desde capilares parapapilares y
tinción de paredes vasculares evidenciando probable vasculitis retiniana.
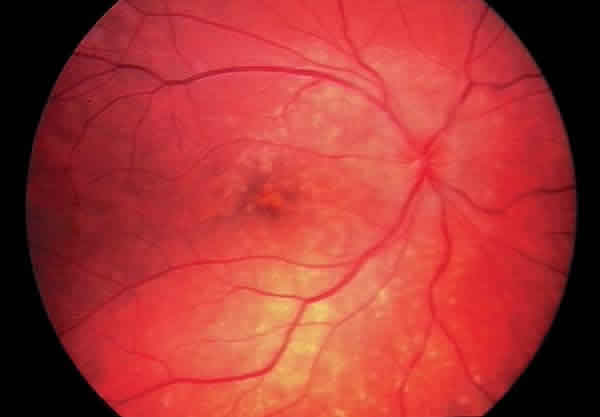
Fig. 2. Retinografía mostrando importante tumefacción papilar y múltiple punteado
blanco peripapilar y siguiendo el inicio de arcadas vasculares, con mayor profusión
a nivel de temporal inferior.
Ante esta clínica, se realiza tratamiento con bolos intravenosos de corticoides: Metil prednisolona (1 gr durante 3 días) , seguido de tratamiento con corticoterapia y ciclosporina oral: Deflazacort (60 mg/día) y Ciclosporina A (200 mg/día).
Se interrumpe la medicación al mes y medio, ante la negatividad de los estudios neurológicos, incluido Resonancia Magnética y negatividad en los datos exploratorios, analíticos y serológicos. Se descartaron enfermedades autoinmunes, infecciosas y desmielinizantes, así como procesos sistémicos que cursan con uveítis. La AV en sucesivas revisiones fue mejorando, estando a los 5 meses en 2/3 y al año en la unidad, manteniéndose así al año y medio. El otro ojo se mantuvo con unidad de AV y sin afectación oftalmoscópica ni angiográfica. Al año, la campimetría, el estudio de potenciales evocados visuales y el electrorretinograma son normales. Pero al año y medio la FAG y el fondo de ojo muestran signos de la afección (figs. 3 y 4).

Fig. 3. Angiografía fluoresceínica correspondiente a la figura 4: hiperfluorescencia
punteada a nivel de la zona primitivamente afecta: mácula y zonas de inicio de
arcadas vasculares, sin alteración papilar, y sin la profusión de estas lesiones, tal
como aparecen en la angiografía de inicio de la enfermedad (fig. 1).

Fig. 4. Retinografía al año y medio que muestra alteración permanente del epitelio
pigmentario dando un patrón granular en mácula, así como en el resto de epitelio
pigmentario primitivamente afectado.
DISCUSIÓN
La dificultad del SMPBE, estriba en su diagnóstico y posible confusión con otras entidades de peor pronóstico funcional (2,5). Especialmente importante por afectar a sujetos jóvenes (1).
Nuestro caso, al principio, se diagnosticó como una coriorretinopatía serosa central. La FAG cambió el diagnóstico a papilitis; la situación de la profusa exudación a neurorretinitis. Esto, acompañado de las lesiones que afectaban al epitelio pigmentario, nos hicieron hacer el diagnóstico diferencial con procesos uveíticos posteriores. La presencia de papilitis sin otros signos, en un joven, nos hizo descartar la esclerosis múltiple.
La evolución final, al año y medio, junto con el conjunto de síntomas, signos y pruebas realizadas nos confirma el diagnóstico de SMPBE (1,2,5).
Comprobamos que existe una gradación en la presentación de la enfermedad. Las secuelas anatómicas, pero no funcionales, muestran una variante con respecto a la descripción primera que de esta enfermedad hizo Jampol (1).
Las lesiones fundoscópicas así como angiográficas, una vez recuperada la función visual, demuestran que ha habido una alteración permanente del epitelio pigmentario. Los signos de vitritis periférica, en la zona primitivamente afectada, nos hablan de una forma crónica y localizada de ruptura de la barrera hematorretiniana. Este estado se mantiene después de año y medio de seguimiento.
Confirmamos con otros autores la posibilidad de presentar lesiones residuales (3,4).
BIBLIOGRAFÍA
1. Jampol LM, Sieving PA, Pugh D, Fishman GA, Gilbert H. Multiple evanescent white dot syndrome. I. Clinical findings. Arch Ophthalmol 1984; 102: 671-674. [ Links ]
2. Arbet TP. Multiple evanescent white dot syndrome. J Am Optom Assoc 1997; 68: 769-774. [ Links ]
3. Wyhinny GJ, Jackson JL, Jampol LM, Caro NC. Subretinal neovascularization following multiple evanescent white dot syndrome. Arch Ophthalmol 1990; 108: 1384. [ Links ]
4. McCollum CJ, Kimble JA. Peripapillary subretinal neovascularization associated with multiple evanescent white dot syndrome. Arch Ophthalmol 1992; 110: 13-14. [ Links ]
5. Verougstraete C. White spots syndromes. Bull Soc Belge Ophtalmol 2001; 279: 67-78. [ Links ]

