










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.80 no.11 nov. 2005
COMUNICACIÓN CORTA
SCA-7. DISTROFIA DE CONOS-BASTONES EN EL SENO DE UNA ATAXIA HEREDITARIA
SCA-7. CONE-ROD DYSTROPHY IN THE CONTEXT OF AN HEREDITARY ATAXIA
ARNALICH-MONTIEL F1, REBOLLEDA G2, MUÑOZ-NEGRETE FJ2
| RESUMEN Caso clínico: Varón de 21 años se presenta con pérdida de visión bilateral desde hace un año, y ataxia cerebelosa desde la infancia. Dos familiares presentaban un cuadro clínico similar. En la exploración se objetivaron escotomas centrales bilaterales y patrón compatible con distrofia conos-bastones en electrorretinograma y tomografía de coherencia óptica. El análisis molecular mediante amplificación por PCR y genotipado del gen SCA7 estableció el diagnóstico de SCA-7, un síndrome genético por expansión de poliglutaminas. Palabras clave: SCA 7, ataxia espinocerebelosa, distrofia conos-bastones, tomografia de coherencia optica, ataxina-7. | ABSTRACT Clinical case: A 21-year-old male presented with bilateral loss of visual acuity within the last year, and cerebellar ataxia since childhood. Two members of his family had a similar disorder. Examination showed bilateral central scotomas, as well as an electroretinogram pattern and optic coherence tomography images consistent with cone-rod dystrophy. Molecular analysis by PCR amplification and genotyping of the SCA7 gene established the diagnosis of SCA-7. Key words: SCA-7, spinocerebellar ataxia, cone-rod dystrophy, optical coherence tomography, ataxin-7. |
Recibido: 26/11/04. Aceptado: 21/10/05.
Servicio de Oftalmología. Unidad de Glaucoma y Neuro-oftalmología. Hospital Ramón y Cajal. Madrid. España.
Universidad de Alcalá de Henares. Alcalá de Henares. Madrid. España.
1 Licenciado en Medicina. Servicio de Oftalmología.
2 Doctor en Medicina. Servicio de Oftalmología. Unidad de Glaucoma.
Comunicación presentada parcialmente en el LXXX Congreso de la S.E.O. (Córdoba 2004).
Correspondencia:
Francisco Arnalich-Montiel
C/. General Kirkpatrick, 27, portal 1, 2.º B
28027 Madrid
España
E-mail: arnalich@hotmail.com
INTRODUCCIÓN
Las alteraciones oculares más frecuentes en los trastornos hereditarios neurodegenerativos conocidos como ataxias espinocerebelosas son las alteraciones oculomotoras, especialmente la diplopía, la oftalmoplejía, o el nistagmo. La pérdida progresiva de la agudeza visual por afectación retiniana es un elemento propio de un subtipo de ataxia, el SCA-7 (1).
Este síndrome, de herencia autosómica dominante, forma parte de un grupo de enfermedades hereditarias que se caracterizan por la expansión inestable de tripletes de nucleótidos, y se presenta como un trastorno de progresión lenta de la coordinación motora, con ataxia, disartria, signos piramidales, que se asocia a una distrofia de conos-bastones.
Se presenta el caso de un varón joven con este síndrome, y se discuten las características y patogenia del mismo.
CASO CLÍNICO
Varón de 21 años, que consulta por disminución de agudeza visual en ambos ojos de un año de evolución, con ataxia cerebelosa progresiva desde la infancia. Como antecedentes familiares, la madre y la abuela materna presentan un cuadro similar.
En la exploración oftalmológica la agudeza visual corregida era 0,2 en ambos ojos, siendo el resto del examen ocular normal.
El campo visual (analizador de campos visuales Humphrey modelo 750®; Zeiss Humphrey Systems; Dublín, California 450) mostraba escotomas centrales bilaterales (estrategia 24-2 SITA).
El patrón del electrorretinograma flash siguiendo la normativa ISCEV (PRIMUS 2,5®; Tomey Corporation, Nagoya, Japón) reveló una distrofia de conos-bastones (fig. 1) con reducción en la amplitud de todas las respuestas, pero más acusada en las respuesta fotópica y flicker (30 Hz).

Fig 1. ERG de un individuo sano (a la derecha) junto a las alteraciones que se encuentran en el paciente afecto de SCA-7 (a la izquierda).
Amplitud reducida en las 5 respuestas, más acentuada en las respuestas dependientes de los conos (fotópica y flicker).
En el examen con tomografía de coherencia óptica (Stratus OCT®, Zeiss Humphrey) se observó un adelgazamiento retiniano foveal y parafoveal (fig. 2) y un área de baja reflectividad en el complejo retina externa-coroides en AO (fig. 3).
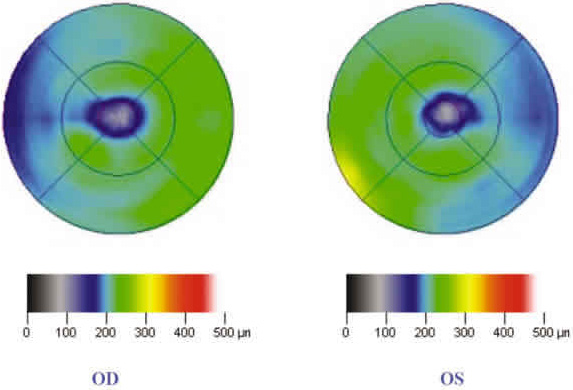
Fig 2. Adelgazamiento en la zona foveal y parafoveal en el mapa topográfico macular del TCO.

Fig 3. TCO: corte-seccional de la retina. Se observa un área de baja reflectividad
que separa el complejo retina externa-coroides.
El estudio genético con PCR estableció el diagnóstico de SCA-7, pues presentaba heterozigosis en el gen que codifica la ataxina 7, localizado en el cromosoma 3, mostrando el alelo afectado 50 repeticiones del triplete de nucleótidos CAG.
DISCUSIÓN
La patogenia de esta enfermedad tiene unas bases genéticas muy interesantes, que comparte con otras enfermedades como el síndrome X Frágil, la enfermedad de Huntington, la distrofia miotónica, y la ataxia de Friedrich (2).
La alteración genética autosómica dominante se debe a la expansión, transmitida de generación en generación, del triplete de nucleótidos CAG (citosina-adenina-guanina), que codifica para el aminoácido glutamina, produciéndose una proteína, la ataxina 7, que contiene un número expandido de poliglutaminas (poli-Q) (2). La expansión de los tripletes es inestable, y al avanzar en el árbol genealógico, se incrementa el número de tripletes CAG. En concreto, la transmisión paterna de la enfermedad produce un mayor incremento del número de repeticiones de trinucleótidos que la transmisión materna (2). Ésta es la base del fenómeno de anticipación por el cual, los descendientes tienen un cuadro más grave y más precoz de la misma enfermedad, pues la proteína es cada vez más anómala. De esta forma, puede darse el caso de que la descendencia manifieste la enfermedad antes incluso de que la manifieste el progenitor, enmascarando así la sospecha de enfermedad hereditaria (1).
La mutación del gen de la ataxina 7, provoca una ganancia de función de la proteína anómala, es decir, que la enfermedad no se debe a la carencia de función de la proteína mutada, sino a la adquisición de nuevas funciones que antes no tenía. Esta nueva función consiste en una activación de las caspasas y la consiguiente inducción de apoptosis neuronal mediada por mitocondrias (3).
Se especula acerca del mecanismo de acción que pudiera explicar por qué solo en este tipo de ataxia se produce la alteración retiniana. Una de las hipótesis propuestas es que se debe a una interacción de la proteína mutada con el factor de transcripción CRX que regula genes específicos de fotorreceptores (homeobox cone-rod), produciéndose la degeneración o distrofia de dichas células (4).
El SCA-7 es, en definitiva, la única ataxia espinocerebelosa conocida que se asocia a la distrofia de conos-bastones (5). Por ello es de gran utilidad la aportación del oftalmólogo en esta patología neurodegenerativa, precisando un estudio electrofisiológico o las imágenes de un tomógrafo de coherencia óptica, para llegar al diagnóstico de distrofia retiniana. Las alteraciones en el ERG y la TCO expuestas en este artículo ya han sido descritas y por tanto contrastadas previamente en la literatura (2).
BIBLIOGRAFÍA
1. Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics and pathogenesis. Lancet Neurol 2004; 3(5): 291-304. [ Links ]
2. Aleman TS, Cideciyan AV, Volpe NJ, Stevanin G, Brice A, Jacobson SG. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res 2002; 74: 737-745. [ Links ]
3. Wang HL, Yeh TH, Chou AH, Kuo YL, Luo LJ, He CY, et al. Polyglutamine-expanded ataxin-7 activates mitochondrial apoptotic pathway of cerebellar neurons by upregulatin Bax and downregulating Bcl-x(L). Cell Signal 2005; Jun 15 (Epub ahead of print). [ Links ]
4. Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder L, et als. Cone-rod dystrophy due to mutation in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997; 91: 543-553. [ Links ]
5. Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet 2004; 12: 2-15. [ Links ]

