










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.81 no.6 jun. 2006
ARTÍCULO ORIGINAL
Distrofia macular corneal: características clínicas, histopatológicas y ultraestructurales
Corneal macular dystrophy: clinical, histopathologic and ultrastructural features
Gulias-Cañizo R.1, Castañeda-Díez R.1, Gómez-Leal A.2, Klintworth G.K.2, Rodríguez-Reyes A.A.3
Servicio de Patología Oftálmica. Asociación para Evitar la Ceguera en México, I.A.P.,
Hospital "Dr. Luis Sánchez Bulnes". México.
1 Doctor en Medicina. Oftalmólogo.
2 Doctor en Medicina. Oftalmólogo. Patólogo Oftálmico.
3 Doctor en Medicina. Anatomopatólogo. Patólogo Oftálmico.
Dirección para correspondencia
RESUMEN
Objetivo: Determinar las principales características clínicas, histopatológicas y ultraestructurales en pacientes mexicanos con distrofia macular de la córnea, así como las alteraciones genéticas en algunos de ellos y compararlas con lo informado en la literatura.
Método: Se recopilaron un total de seis casos con diagnóstico histopatológico de distrofia macular corneal registrados de 1957 a 2004.
Resultados: Clínicamente todas las córneas presentaban opacidades estromales blanco grisáceas de bordes difusos. Histopatológicamente se demostró la presencia de depósitos finamente granulares entre las laminillas del estroma corneal, que resultaron intensamente positivos con las tinciones de azul alciano e hierro coloidal. Por microscopía electrónica de transmisión se demostró dilatación del retículo endoplásmico liso y la presencia de vacuolas intracitoplásmáticas con un material electrodenso, correspondiente a glicosaminoglicanos. El análisis genético en dos de los pacientes demostró mutaciones en el gen CHST6.
Conclusiones: En nuestra serie hubo un predominio de afección por el sexo femenino y el promedio de edad fue mayor en comparación a lo informado en la literatura. Los hallazgos clínicos, histopatológicos y ultraestructurales fueron similares a aquellos previamente informados en otras series. Al igual que en la literatura, la alteración genética en 2 de nuestros pacientes se encontró en el gen CHST6.
Palabras clave: Córnea, corneal, distrofias corneales, distrofia macular, distrofias estromales.
ABSTRACT
Objective: To assess the main clinical, genetic, histopathological and ultrastructural features of Mexican patients with macular corneal dystrophy, and to compare the results with those previously reported.
Method:We analyzed six cases where a histopathologic diagnosis of macular corneal dystrophy had been made between 1957 and 2004.
Results: Clinically, all corneas showed focal grayish-white stromal opacities with diffuse edges. Histopathologically, intrastromal granules stained strongly positive with Alcian blue and colloidal iron. Transmission electron microscopy showed enlargement of smooth endoplasmic reticulum and the presence of intracytoplasmic vacuoles that corresponded to glycosaminoglycans. Genetic analysis showed novel mutations in the CHST6 gene in 2 of the patients.
Conclusions:Females were more affected than males and the mean age at the time of diagnosis was older than that reported previously, however the clinical, histopathological and ultrastructural features were similar to those of previous reports. As described in other cases in the literature, in some instances a disorder is found in CHST6 gene as a basis for this condition. (Arch Soc Esp Oftalmol 2006; 81: 315-320).
Key words: Cornea, corneal, corneal dystrophy, macular dystrophy, stromal dystrophies.
Introducción
La distrofia macular de la córnea (DMC) es un trastorno autosómico recesivo que presenta una alteración en el metabolismo del queratán sulfato. De las distrofias estromales es la menos frecuente, pero la que da manifestaciones a una edad más temprana. Esta distrofia no se asocia con anomalías sistémicas, sin embargo, hay un caso clínico publicado de un paciente con esta entidad asociada a hipotricosis (1).
Se han descrito dos tipos que son indistinguibles clínica e histológicamente. El tipo I caracterizado por ausencia de queratán sulfato sulfatado en córnea, suero y cartílago. El tipo II presenta queratán sulfato sulfatado en estas mismas localizaciones pero con una síntesis de 30% menor a lo normal.
El cuadro clínico en estos pacientes consiste en una córnea clara al nacimiento que comienza a opacificarse a partir de los 3 años de edad. Se observa la presencia de opacidades estromales focales de color blanco grisáceo que al inicio de la enfermedad son superficiales y centrales pero a medida que avanza la misma se extienden hacia el estroma profundo y a la periferia corneal. Puede existir afección endotelial.
Los síntomas principales de esta distrofia consisten en disminución de la agudeza visual y fotofobia.
Histopatológicamente se caracteriza por la presencia de depósitos extracelulares de mucosustancias ácidas que corresponden a glicosaminoglicanos (GAG), tanto entre las lamelas estromales como en el citoplasma de las células endoteliales. Estos depósitos son intensamente positivos con las tinciones de azul alciano y hierro coloidal. Por ultraestructura se observa dilatación del retículo endoplásmico liso y formación de vacuolas intracitoplasmáticas debido a la presencia de un material electrodenso que corresponde a GAG.
Se han descrito mutaciones en el nivel del cromosoma 16q22, en un gen llamado CHST6, que codifica la enzima corneal glucosamina N-acetil-6 sulfotransferasa (C-GlcNac-6-ST), la cual funciona como sulfotransferasa de carbohidratos. (2)
El tratamiento para estos pacientes en etapas avanzadas de la enfermedad, consiste en una queratoplastía penetrante (3).
El presente trabajo tiene como principal objetivo, determinar las principales características clínicas, histopatológicas y ultraestructurales de pacientes Mexicanos con DMC así como las alteraciones genéticas en algunos de ellos y compararlas con las informadas en la literatura.
Sujetos, Material y Métodos
Se revisaron todos los casos con diagnóstico histopatológico de DMC registrados en un período comprendido entre 1957 y 2004.
De todos los casos se realizaron cortes de 5 micras de tejido fijado en formol al 10% e incluido en parafina, los cuales fueron teñidos con hematoxilina y eosina, ácido peryódico de Schiff, azul alciano y hierro coloidal.
En uno de los casos se realizaron además cortes semifinos, se tiñeron con azul de toluidina y se seleccionaron ciertas áreas específicas para el estudio de microscopia electrónica de transmisión, que se efectuó mediante la técnica convencional.
En dos de los pacientes se tomaron 30 ml de sangre venosa periférica, de 10 ml de cada uno de ellos se extrajo el ADN utilizando un kit de aislamiento. El ADN extraído junto con los 20 ml restantes de sangre fresca de cada paciente, fueron enviados al Centro Médico de la Universidad de Duke en Durham, Caroliona del Norte, donde uno de los autores (GKK) junto con su equipo de trabajo se encargaron de la realización del análisis genético. La toma de la muestra sanguínea se obtuvo con el consentimiento informado previamente firmado por cada uno de los pacientes participantes.
Resultados
Los principales hallazgos clínicos se resumen en la tabla I. Se recopilaron un total de seis pacientes, cuatro mujeres y dos hombres, con un intervalo de edad entre los 24 y los 41 años y un promedio de edad al momento del diagnóstico de 32,5 años.
La afección fue bilateral en todos los casos y clínicamente se manifestó de manera muy similar en todos ellos, caracterizada por la presencia de opacidades corneales de 0,5 a 1 mm de eje mayor, de color blanco grisáceo y de bordes difusos, que involucraban todo el espesor estromal, predominantemente el tercio superficial, así como el endotelio. El estroma localizado entre las opacidades presentaba un velamiento difuso (fig. 1). El tiempo de evolución fue largo en la mitad de los casos y fluctuó entre los 5 y 14 años. En la otra mitad no fue especificado. El diagnóstico clínico fue de distrofia macular en cuatro pacientes, de distrofia granular en un paciente y de amiloidosis subepitelial familiar en el paciente restante.
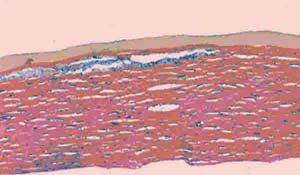
Fig. 1. Aspecto clínico que muestra opacidades
corneales difusas de bordes mal definidos.
El tratamiento de elección en todos los pacientes fue la queratoplastía penetrante, en cuatro de ellos en forma unilateral y en dos en forma bilateral. Ninguno de los pacientes presentó recurrencia en un tiempo de seguimiento que varió entre 3 y 9 años posterior al transplante corneal.
Histopatológicamente todas las córneas presentaron depósitos de un material finamente granular entre las laminillas del estroma corneal, con un predominio en el tercio superficial. Estos mismos depósitos estaban presentes en el citoplasma de las células endoteliales. Los depósitos resultaron intensamente positivos con las tinciones de azul alciano y hierro coloidal (fig. 2). No se realizaron inmunotinciones en ninguno de los casos por no contar con los anticuerpos para proteoglicanos y glicosaminoglicanos. Por ultraestructura se demostró dilatación del retículo endoplásmico liso y la presencia de vacuolas intracitoplasmáticas con un material electrodenso, correspondiente a GAG (fig. 3).

Fig. 2. Depósitos intraestromales finamente granulares
predominantemente en el tercio superficial
(Hierro coloidal, ampliación original x10).

Fig. 3. Electromicrografía que muestra grandes vacuolas
intracitoplasmáticas ocupadas por material electrodenso
(glicosaminoglicanos).
En el análisis genético, en uno de los pacientes se encontraron dos mutaciones, mientras que en otro paciente solo se encontró una mutación (tabla II). No se encontraron alteraciones genéticas en otra región fuera del gen CHST6.

Discusión
En nuestra serie de casos de DMC hubo un predominio de afección por el género femenino, a diferencia de lo informado en otras series en las que no se ha demostrado un predominio de afección por sexo.
El promedio de edad de nuestros pacientes al momento del diagnóstico fue de 32,5 años, mayor a lo informado en la literatura. Creemos que la explicación puede deberse a que los pacientes de nuestra serie con esta enfermedad provienen de comunidades rurales alejadas de las grandes ciudades, por lo que acuden a la consulta en estadios tardíos de la enfermedad, cuando ésta se hace sintomática, causando como principal repercusión una disminución de la agudeza visual.
Debido a que esta distrofia se hereda con patrón de transmisión autosómico recesivo, es de esperarse que cada individuo afectado tenga dos mutaciones en el gen CHST6. De nuestros dos pacientes estudiados, en uno de ellos se encontró una sola mutación en este gen. Si bien las mutaciones de novo y la deleción encontradas no han sido previamente informadas, éstas sí se localizan en la región del gen CHST6. En el estudio realizado por Akama et al (2). Se encontraron varias mutaciones así como deleciones y/o reemplazos largos en la región que codifica para el gen CHST6. En otro estudio realizado por El-Ashry et al (4), se identificaron 6 mutaciones de novo en 5 familias distintas. Por otro parte, en un estudio de Aldave et al (5) se identificaron 4 mutaciones distintas en el mismo gen. Los resultados de estos estudios, aunados a los nuestros, demuestran que son varias las alteraciones genéticas que pueden encontrarse en la región que codifica para el gen CHST6, ya sean mutaciones de novo (homocigotos o heterocigotos), deleciones y/o reemplazos. Sin embargo, hasta el momento actual, los pacientes con DMC tienen características clínicas e histológicas indistinguibles independientemente del sustrato genético que presenten. Diversos informes en la literatura describen la presencia de homocigotos en esta enfermedad, probablemente por la dificultad técnica que conlleva encontrar ambas mutaciones (4).
Akama et al (2) analizaron el RNA mensajero en córneas humanas y demostraron que la expresión normal de este gen se correlaciona con la presencia de queratán sulfato sulfatado en la córnea, característica normal que promueve la organización laminar y por lo tanto la transparencia corneal. Por otro lado, Hasegawa et al (6) demostraron que una disminución en la actividad de la C-GlcNac-6-ST (secundaria a una expresión anormal del gen CHST6) se relaciona con la presencia de queratán sulfato no sulfatado en córneas humanas, y la presencia del mismo causa opacidades corneales. En resumen, estas anomalías genéticas son las responsables de una pérdida en la función de la sulfotransferasa de carbohidratos, alteración responsable del fenotipo de la distrofia macular de la córnea secundariamente a la presencia de queratán sulfato no sulfatado, lo que conlleva a una pérdida de la organización y estructura normal de la sustancia del estroma corneal.
Aunque se ha informado recurrencia de la distrofia en el injerto, la tasa de la misma es la más baja comparada con las otras distrofias estromales (7-10). Ninguno de nuestros casos presentó recurrencia en un tiempo de seguimiento hasta de 9 años posteriores al transplante corneal. Kuchle et al (7) informaron un caso con recurrencia de DMC tipo II, 49 años después de una queratoplastía penetrante.
Hasta dónde es de nuestro conocimiento, esta corresponde a la primera serie de casos de pacientes mexicanos con DMC informada en Latinoamérica, con una mayor incidencia de afección en mujeres, con un promedio de edad mayor y sin recurrencia de la enfermedad en algunos casos hasta 9 años después de la queratoplastía penetrante. La presentación clínica, los hallazgos histopatológicos y ultraestructurales, así como las alteraciones genéticas son muy similares a aquellas previamente informadas en otras series.
Bibliografía
1. Becker M, Rohrschneider K, Tilgen W, Weber BH, Volcker HE. Familial juvenile macular dystrophy with congenital hypotrichosis capitis. Ophthalmologe 1998; 95: 233-240. [ Links ]
2. Akama TO, Nishida K, Nakayama J, Watanabe H, Ozaki K, Nakamura T et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet 2000; 26: 237-241. [ Links ]
3. Klintworth GK, Reed J, Stainer GA, Binder PS. Recurrence of macular corneal dystrophy within grafts. Am J Ophthalmol 1983; 95: 60-72. [ Links ]
4. El-Ashry MF, El-Aziz MM, Wilkins S, Cheetham ME, Wilkie SE, Hardcastle AJ et al. Identification of novel mutations in the carbohydrate sulfotransferase gene (CHST6) causing macular corneal dystrophy. Invest Ophthalmol Vis Sci 2002; 43: 377-382. [ Links ]
5. Aldave AJ, Yellore VS, Thonar EJ, Udar N, Warren JF, Yoon MK et al. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am J Ophthalmol 2004; 137: 465-473. [ Links ]
6. Hasegawa N, Torii T, Kato T, Miyajima H, Furuhata A, Nakayasu K et al. Decreased GlcNAc 6-O-sulfotransferase activity in the cornea with macular corneal dystrophy. Invest Ophthalmol Vis Sci 2000; 41: 3670-3677. [ Links ]
7. Kuchle M, Cursiefen C, Fischer DC, Shlotzer-Schrehardt U, Nauman GO. Recurrent macular corneal dystrophy type II 49 years after penetrating keratoplasty. Arch Ophthalmol 1999; 117: 528-531. [ Links ]
8. Davies Y, Fullwood NJ, Marcyniuk B, Bonshek R, Tullo A, Nieduszynski IA. Keratan sulphate in the trabecular meshwork and cornea. Curr Eye Res 1997; 16: 677-686. [ Links ]
9. Lang GK, Naumann GO. The frequency of corneal dystrophies requiring keratoplasty in Europe and the U.S.A. Cornea 1987; 6: 209-211. [ Links ]
10. Edward DP, Thonar EJ, Srinivasan M, Yue BJ, Tso MO. Macular dystrophy of the cornea. A systemic disorder of keratan sulfate metabolism. Ophthalmology 1990; 97: 1194-1200. [ Links ]
![]() Dirección para correspondencia
Dirección para correspondencia
Rosario Gulias-Cañizo
Vicente García Torres #46, Col. San Lucas Coyoacán
Servicio de Patología Oftálmica
México
E-mail: bichorro76@hotmail.com
Recibido: 14.9.05.
Aceptado: 29.5.06.

