










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.81 no.6 jun. 2006
COMUNICACIÓN CORTA
Síndrome de Weill-Marchesani: afectación familiar
Weill-Marchesani's syndrome: familial involvement
Veiga de la Jara C.1, Bosch Valero J.2, Torres Suárez E.1, Mateos Sánchez E.1,
Rojo Castejón P.3, Ancochea Díaz G.1
Hospital Universitario Ramón y Cajal. Madrid. España.
1 Licenciado en Medicina.
2 Licenciado en Medicina. Hospital Clínico Lozano Blesa.
3 Doctora en Medicina.
Comunicación presentada en el LXXVIII Congreso de la S.E.O. (Murcia 2002).
Dirección para correspondencia
RESUMEN
Caso clínico: Presentamos el caso clínico de un niño con talla baja, braquidactilia y braquimorfia remitido a la consulta por baja visión. Se trata de un caso de síndrome de Weill-Marchesani en una familia en la que cuatro de los ocho hijos presentan esferofaquia, braquimorfia y braquidactilia.
Discusión: Existen pocos casos familiares del síndrome de Weill-Marchesani reflejados en la literatura. Se han descrito patrones de herencia tanto autosómico dominante como recesivo. El papel del oftalmólogo en su diagnóstico y manejo es esencial, ya que la afectación oftalmológica es la más grave.
Palabras clave: Síndrome de Weill-Marchesani, afectación familiar, ectopia lentis, esferofaquia.
ABSTRACT
Case report: We report the case of a child short in stature with brachydactyly and brachymorphy who was referred to our office complaining of poor vision. This was a case of Weill-Marchesanis syndrome described in a family, in which four of the eight children were affected by spherophakia, brachymorphy and brachydactyly.
Discussion: There are few familial cases of Weill-Marchesanis syndrome reported in the literature. Both autosomal dominant and recessive inheritances have been described. The opththalmologist plays a crucial role in its diagnosis and management, since the ocular involvement is the most severe one. (Arch Soc Esp Oftalmol 2006; 81: 349-352).
Key words: Weill-Marchesanis syndrome, familial involvement, ectopia lentis, spherophakia.
Introducción
El síndrome de Weill-Marchesani es una alteración poco frecuente del tejido conectivo con afectación ocular. Los pacientes presentan baja talla, braquidactilia con rigidez articular, microsferofaquia, miopía lenticular progresiva, luxación cristaliniana y glaucoma secundario. Desde su descripción por Weill y Marchesani en 1932 y 1939 (1) se han documentado patrones de herencia autosómica dominante y recesiva (1-3).
Caso clínico
Un varón de 13 años es remitido para valoración oftalmológica por mala visión. Fenotípicamente era un niño de talla corta con una estatura de 133 cm, braquimorfia y braquidactilia en las cuatro extremidades (figs. 1 y 2).
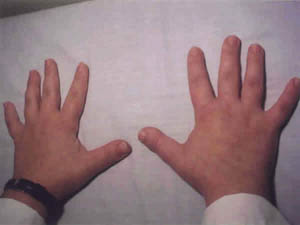
Fig. 1. Manos cortas y braquidactilia.
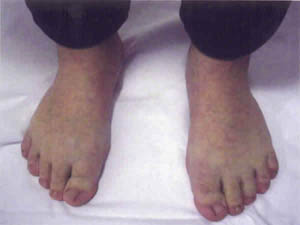
Fig. 2. Braquidactilia en extremidades inferiores.
El paciente presentaba un error refractivo corregido de –13,00 –6,50 a 1º en el ojo derecho y de –16,00-6,25 a 179º en el izquierdo. Con dicha corrección alcanzaba una agudeza visual de 0,4 y 0,2 respectivamente. No existía diplopía monocular ni hallazgos en la motilidad ocular extrínseca e intrínseca.
El diámetro corneal horizontal era de 12,0 mm en ambos ojos y la paquimetría de 613 y 611 micras respectivamente. La cámara anterior era estrecha, apreciándose iridofacodonesis bilateral. Se evidenció microesferofaquia con desplazamiento anterior de ambos cristalinos dentro de la cámara posterior (fig. 3).
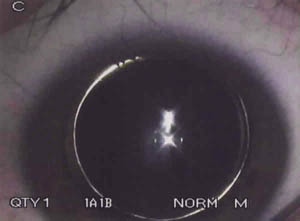
Fig. 3. Esferofaquia en la que el diámetro
anteroposterior del cristalino es grande con
respecto al diámetro a nivel del ecuador.
La presión intraocular era de 20 mmHg bilateralmente. Gonioscópicamente se apreció un ángulo estrecho simétrico en ambos ojos grado II según Schaffer.
La exploración mediante topógrafo Orbscan-II (Topógrafo corneal, Baush and Lomb, U.S.A.) y el examen del fondo de ojo no mostraron alteraciones.
Como prevención del cierre angular se realizó una iridotomía láser Nd:YAG en ambos ojos a las 10 y 2 horas respectivamente. Dada la estabilidad de la visión y la ausencia de diplopía se adoptó una actitud conservadora de acuerdo con el deseo de la familia del paciente mediante control de la agudeza visual, presión intraocular, documentación fotográfica y la realización de campos visuales para detectar la aparición de daño glaucomatoso.
Revisando sus antecentes familiares se identificó al paciente como el menor de una familia de ocho hermanos, en la que además del mencionado un hermano y dos hermanas presentaban esferofaquia, braquimorfia y braquidactilia (fig. 4). Los padres eran sanos y ninguno de los hijos ha tenido descendencia.

Fig. 4. Miembros de la familia del probando,
en la que los tres hermanos situados a su
derecha están afectos.
Discusión
El diagnóstico clínico del síndrome de Weill-Marchesani se basa en las anomalías cristalinianas y esqueléticas. Además de la corta estatura, braquidactilia y braquimorfia, pueden encontrarse alteraciones oculares como la miopía lenticular, microesferofaquia, ectopia lentis y glaucoma secundario (2). Actualmente se considera que la esferofaquia no es imprescindible para el diagnóstico (1). Otros hallazgos menos frecuentes son la licuefacción vítrea, degeneración coriorretiniana, megalocórnea y microcórnea (1).
Un cristalino normal es casi esférico al nacimiento, con un diámetro ecuatorial de 6,5 mm y un diámetro anteroposterior (sagital) de 3,5 a 4,0 mm. El diámetro ecuatorial crece rápidamente hasta alcanzar los 9,0 mm a los 15 años y permanece así durante el resto de la vida. Por el contrario, el diámetro sagital es de unos 3,7 mm a los 20 años y de 4,0 a los 50 años de edad, alcanzando los 4,75-5,0 mm a los 80-90 años de edad. La mayor parte de los cambios fisiológicos cristalinianos ocurren en la superficie anterior del cristalino, el cual se torna más esférico a medida que envejece (2).
En la microsferofaquia el cristalino es pequeño y de forma esférica. La causa de la microsferofaquia es desconocida, aunque se ha propuesto que es debida a una alteración en el desarrollo de las fibras cristalinianas secundarias y/o a un defecto en la inserción de las fibras secundarias anómalamente finas. Otras teorías atribuyen la microsferofaquia a la falta de tensión en sus fibras zonulares rudimentarias, impidiendo el desarrollo fisiológico del cristalino y ocasionando su forma esférica. El diámetro sagital del cristalino normal cambia poco durante la infancia y pubertad, ya que la adecuada tensión en las fibras zonulares limitan la expansión en el plano sagital. Una anomalía en las fibras zonulares permitiría al cristalino permanecer esférico en lugar de tender a una forma biconvexa con el paso de los años (1). Todo ello, ligado a la tendencia a la luxación y desplazamiento del cristalino explicarían la progresión de la miopía lenticular.
La miopía significativa aparece a la edad media de 11,2 años, la subluxación cristaliniana sucede a los 18,2 años, y su desplazamiento suele ocurrir inferiormente, al contrario que en el síndrome de Marfan y la homocistinuria (2). En nuestro paciente de 13 años se objetiva miopía y ectopia del cristalino, aunque el desplazamiento cristaliniano es en sentido anterior.
La pérdida de visión obedece a la miopía y al daño glaucomatoso por estrechamiento progresivo de la cámara anterior y cierre angular (2).
Las indicaciones para la cirugía del cristalino son su dislocación a cámara anterior, subluxación progresiva con bisección pupilar, dislocación inminente, diplopía monocular y agudeza visual menor de 20/70 para lejos y de J5 para cerca (4).
El manejo actual comprende la extracción cristaliniana con o sin colocación de lente intraocular y vitrectomía anterior (4), además del tratamiento médico o quirúrgico del glaucoma secundario. No obstante existe un mayor riesgo de complicaciones postquirúrgicas en estos pacientes, como atalamia tras cirugía filtrante (5) o roturas zonulares (2,5).
Todavía no ha sido aclarada la presunta disparidad entre el síndrome de Weill-Marchesani con patrones de herencia dominante y recesiva. Tradicionalmente la ectopía cristaliniana fue considerada un rasgo autosómico recesivo, aunque existen casos documentados de herencia autosómica dominante. Se ha sugerido el acrónimo inglés GEMSS (glaucoma, ectopia lentis, microsferofaquia, rigidez articular, baja estatura) para describir la forma dominante, que presenta una posible relación con el síndrome de Moore-Federman, siendo la ectopía cristaliniana el rasgo diferencial entre ambas entidades (3). La escasa documentación sobre familias afectadas hace necesaria un mayor número para dilucidar la transmisión de este síndrome. En la literatura española consultada (Medline) no hemos encontrado referencia a ninguna familia con más de un caso de síndrome de Weill-Marchesani.
Como ya se ha postulado en este caso el gen podría ser dominante con una reducida penetrancia y un amplio rango de expresividad por el hallazgo de talla baja, braquidactilia y esferofaquia en muchos miembros de la familia. Otra alternativa sería la naturaleza incompleta recesiva del gen, con expresión parcial en heterocigotos (1,3). Dada la baja estatura del padre no es posible excluir la pseudodominancia como patrón de herencia.
Wirtz ha sugerido que el 15q21.1. sería el gen responsable del síndrome de Weill-Marchesani (3).
El oftalmólogo desempeña una labor diagnóstica y pronóstica esencial en patologías sistémicas con afectación ocular como el síndrome de Weill-Marchesani. La corrección óptica precoz previene la ambliopía y el seguimiento del paciente permite detectar y tratar el glaucoma secundario.
Bibliografía
1. Evereklioglu C, Hepsen IF, Er H. Weill-Marchesani syndrome in three generations. Eye 1999; 13: 773-777. [ Links ]
2. Jensen AD, Cross HE, Paton D. Ocular complications in the Weill-Marchesani syndrome. Am J Ophthalmol 1974; 77: 261-269. [ Links ]
3. Wirtz MK, Samples JR, Kramer PL, Rust K, Yount J, Acott TS et al. Weill-Marchesani syndrome--possible linkage of the autosomal dominant form to 15q21.1. Am J Med Genet 1996; 65: 68-75. [ Links ]
4. Halpert M, BenEzra D. Surgery of the hereditary subluxated lens in children. Ophthalmology 1996; 103: 681-686. [ Links ]
5. Maquet Dusart JA, Dios Castro E. Síndrome de Weill-Marchesani. Atalamia tardía tras cirugía antiglaucomatosa. Arch Soc Esp Oftalmol 2002; 77: 385-387. [ Links ]
![]() Dirección para correspondencia
Dirección para correspondencia
Carlos Veiga de la Jara
Servicio de Oftalmología. Secretaría de Dacriología.
Hospital Ramón y Cajal
Ctra. de Colmenar, km. 9,100.
28034 Madrid
España
E-mail: veigajara@hotmail.com
Recibido: 22.2.05.
Aceptado: 19.5.06.

