










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.81 no.7 jul. 2006
ARTÍCULO ORIGINAL
Distrofia corneal granular autosómica dominante causada por mutación del gen TGFBI en una familia mexicana
Autosomal dominant granular corneal dystrophy caused by a TGFBI gene mutation in a mexican family
Zenteno J.C.1, Santacruz-Valdés C.2, Ramírez-Miranda A.1
Instituto de Oftalmología Conde de Valenciana. México D.F. México.
1 Doctor en Ciencias. Departamento de Genética y Unidad de Investigación.
2 Doctor en Medicina. Departamento de Córnea.
Dirección para correspondencia
RESUMEN
Objetivo: Las distrofias corneales son un grupo de alteraciones hereditarias en las que una acumulación progresiva de material amiloide, hialino o mixto en las distintas capas corneales produce disminución de la transparencia corneal. Se describen las características clínicas y los estudios moleculares del gen TGFBI en una paciente Mexicana con una distrofia corneal estromal de tipo granular.
Métodos: Examen oftalmológico completo, caracterización fenotípica de la distrofia corneal, y análisis del gen TGFBI por reacción en cadena de la polimerasa (PCR) y por secuenciación nucleotídica, en DNA de la propósita y de un hermano afectado.
Resultados: Las lesiones corneales observadas en la paciente fueron compatibles con el diagnóstico de distrofia corneal estromal de tipo granular (clásica). No se observaron lesiones en las otras capas corneales. El análisis del gen TGFBI en DNA de la paciente y de un hermano afectado reveló una mutación puntual, de adenina a guanina, en el exón 14 de TGFBI que origina un cambio de histidina a arginina en el aminoácido 626 (H626R) de la proteína TGFBI.
Conclusiones: Éste es el primer caso en el que se demuestra que una distrofia corneal granular es causada por la mutación H626R en TGFBI. Esta mutación ha sido reportada consistentemente en la distrofia estromal de tipo empalizada, clínicamente diferente a la granular. Nuestros datos indican que existen excepciones en la aparente correlación genotipo-fenoitipo establecida en el grupo de distrofias corneales asociadas a mutación en el gen TGFBI.
Palabras clave: Distrofia corneal, distrofia estromal, enfermedad corneal hereditaria, mutación del gen TGFBI.
ABSTRACT
Objective: To describe the clinical data and the results of molecular analyses of the TGFBI gene in a patient with classic granular stromal corneal dystrophy (type I).
Methods: A female patient aged 60-years complaining of a long-standing decrease of visual acuity bilaterally associated with photophobia and foreign body sensation, underwent a complete ophthalmologic examination. Molecular analyses of DNA from the patient and from an affected brother included PCR amplification of exons 4, 11, 12, and 14 of the TGFBI gene and direct automated sequencing of the PCR products.
Results: The affected patient showed a pattern of corneal stromal lesions that was compatible with a diagnosis of classic granular dystrophy. No involvement of other corneal layers was evident. Molecular analysis disclosed a point mutation in exon 14 of the TGFBI gene which consisted of an adenine to guanine change at nucleotide position 1924, predicting a substitution of arginine instead of histidine at residue 626 of the TGFBI protein (H626R). An identical mutation was detected in DNA from her affected brother.
Conclusions: This is the first time that a case of stromal granular dystrophy has been demonstrated to be caused by the H626R mutation, a molecular defect classically detected in the phenotypically distinct lattice corneal dystrophy. Our data indicate that the same molecular defects in the TGFBI gene lead to different phenotypes in stromal dystrophies, thus expanding the genotypic-phenotypic spectrum in this group of corneal diseases (Arch Soc Esp Oftalmol 2006; 81: 369-374).
Key words: Corneal dystrophy; stromal dystrophy; corneal genetic disease; TGFBI mutation.
Introducción
Las distrofias corneales son un grupo de enfermedades hereditarias caracterizadas por pérdida de la transparencia corneal causada por acumulación progresiva de depósitos anormales en las diferentes capas de la córnea. Estas alteraciones tienen una gran heterogeneidad clínica y genética, inician en las primeras décadas de la vida, afectan principalmente la parte central de la córnea y no se asocian a procesos inflamatorios (1). Con el tiempo, los depósitos llevan a alteración visual y frecuentemente se requieren procedimientos quirúrgicos como queratoplastías penetrantes para restablecer la agudeza visual. La clasificación actual de las distrofias corneales se basa en la capa de la córnea que está afectada y en las características biomicroscópicas de los depósitos, que pueden ser de tipo amiloide, hialino o mixto (2). La mayoría de las distrofias corneales se heredan como rasgos autosómicos dominantes con variabilidad en la expresión clínica y alto grado de penetrancia (3). En años recientes, el estudio de las bases moleculares de diferentes tipos de distrofias corneales ha permitido el reconocimiento de un grupo de distrofias estromales que tienen como origen común mutaciones en el gen TGFBI (Transforming Growth Factor, Beta-Induced) (también conocido como BIGH3), localizado en la región cromosómica 5q31 (2, 4-6). Mutaciones definidas en este gen se correlacionan uniformemente con tipos específicos de distrofias que afectan selectivamente el estroma corneal. A la fecha, se han demostrado mutaciones de TGFBI en pacientes de diversos grupos étnicos con cuatro diferentes distrofias corneales estromales autosómicas dominantes: distrofia corneal granular tipo I (6-8), distrofia corneal granular tipo II o tipo Avellino (6,8,9), distrofia corneal granular tipo III o tipo Reis-Bucklers (8,10,11) y distrofia corneal en empalizada (lattice) de tipos I, IIIA, I/IIIA, IIIB y IV (12-14). Todas las distrofias corneales causadas por mutaciones en TGFBI se caracterizan por un depósito extracelular anormal de proteínas TGFBI mutantes en el estroma corneal (15).
La distrofia corneal granular tipo I, también conocida como distrofia corneal granular clásica, se caracteriza por opacidades múltiples discretas en la córnea que semejan migajas y es el tipo más común de distrofia estromal (16). La alteración se hace evidente generalmente en la primera o segunda década de la vida o en la pubertad con la aparición de opacidades blanco-grisáceas que afectan a la capa estromal superficial de la córnea (17). Con el tiempo, las lesiones tienden a aumentar de tamaño, se agregan y se extienden mas profundamente y hacia la periferia. Típicamente se conserva una zona clara alrededor del limbo corneoescleral y durante la cuarta década de la vida pueden aparecer opacidades centrales en forma de disco. Los sujetos afectados pueden mantener visión normal por largo tiempo ya que la agudeza visual disminuye gradualmente (18).
Se ha observado una correlación fenotipo-genotipo uniforme en la distrofia corneal granular tipo I ya que la gran mayoría de los sujetos con esta distrofia tienen una mutación de citosina (C) a timina (T) en la base 1710 del exón 12 del gen TGFBI, que origina un cambio de arginina a triptofano en el aminoácido 555 de la proteína TGFBI (2,6).
En este trabajo se describe el análisis molecular del gen TGFBI en una familia mexicana con distrofia corneal granular tipo I, de transmisión autosómica dominante y causada por una mutación en el gen TGFBI. Éste es la primera descripción de una mutación en TGFBI en sujetos mexicanos con distrofia corneal granular hereditaria y nuestros resultados permiten expandir el espectro mutacional asociado a las distrofias granulares estromales.
Sujetos, Material y Métodos
El caso índice es una paciente de 60 años (BSA) de edad quien acudió a consulta por disminución progresiva de la agudeza visual en ambos ojos desde la edad de 38 años. La exploración oftalmológica reveló una agudeza visual de 20/200 en ambos ojos con refracción diferida por la presencia de opacidades cornéales importantes de predominio central, que afectaban estroma anterior y medio, con espacios claros entre ellas y que respetaban el limbo corneoescleral (fig. 1); las capas epitelial y endotelial de la cornea exhibieron características normales. La PIO fue de 14 mmHg en ambos ojos, los movimientos oculares no presentaron alteraciones y no se observaron anomalías a la exploración de fondo de ojo. La sintomatología ocular asociada incluyó fotofobia, sensación de cuerpo extraño y percepción de halos alrededor de las luces. No se observaron dismorfias faciales ni malformaciones somáticas asociadas. La historia familiar y el análisis genealógico revelaron que el padre (fallecido), un hermano y una tía paterna habían sido evaluados oftalmológicamente con anterioridad y diagnosticados con «degeneración corneal», y que la transmisión hereditaria de la enfermedad en la familia seguía un patrón autosómico dominante (fig. 2). Aunque los familiares afectados no fueron evaluados directamente por nosotros, se obtuvo muestra de DNA de uno de ellos (hermano) para el estudio genético y confirmación del status de enfermo.
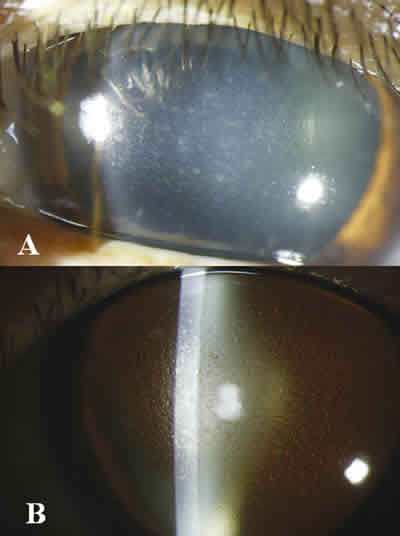
Fig. 1. Fenotipo de la distrofia corneal en la paciente, por
iluminación directa (A) y por retroiluminación (B). Se observan
opacidades granulares finas en estroma que afectan principalmente
la parte central de la córnea y respetan el limbo corneoescleral.
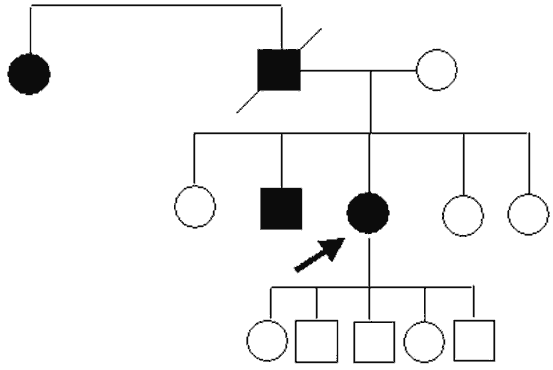
Fig. 2. Árbol genealógico simplificado de la familia con distrofia corneal granular.
Se advierte una transmisión autosómica dominante de la enfermedad. La flecha señala
el caso índice. Los cuadros y círculos indican varones y mujeres, respectivamente;
los símbolos negros indican sujetos con la distrofia corneal.
Estudios moleculares del gen TGFBI: Después de obtener la aprobación de los pacientes y del comité institucional de ética, se extrajo DNA genómico a partir de leucocitos de una muestra de sangre periférica de la propósita y de un hermano, utilizando técnicas estándar. Se realizó la amplificación por reacción en cadena de la polimerasa (PCR) de los exones 4, 11, 12 y 14 del gen TGFBI empleando 4 pares de oligonucleótidos específicos descritos previamente (6). Los productos amplificados por PCR fueron analizados en geles de agarosa al 1,5% de donde las bandas con los productos amplificados fueron cortadas y purificadas utilizando el kit Qiaex II (Qiagen, Charlesworth, USA). Se realizó secuenciación nucleotídica automatizada de los productos de PCR purificados por el método de terminadores ddNTPs marcados con fluorescencia (Applied Biosystems, Foster City, USA). Todas las muestras fueron analizadas en un secuenciador de DNA Genetic Analyzer 310 (Applied Biosystems). Las variaciones de secuencia fueron verificadas en ambas cadenas del DNA. Como DNA control se analizaron 60 alelos del gen TGFBI de sujetos mexicanos sanos (n=30). Las secuencias nucleotídicas fueron comparadas con la secuencia normal del gen TGFBI publicada en la base de datos Ensembl (ENST00000305126).
Resultados
Después de realizar la secuenciación de bases nucleótidicas correspondientes a los exones 4, 11, 12 y 14 del gen TGFBI en DNA de la paciente, se identificó una mutación puntual en el exón 14 en heterozigosis, que consistió en el cambio de una adenina hacia una guanina en el nucleótido 1924. Esta mutación origina una sustitución del codón normal CAT, que codifica el aminoácido histidina en la posición 626 de la proteína, por un codón CGT, que codifica una arginina (fig. 3). El análisis en DNA del hermano afectado demostró un cambio idéntico al de la propósita confirmando el carácter familiar de la mutación y de la distrofia corneal. Esta mutación no fue detectada en ninguno de los 60 alelos de sujetos mexicanos normales.

Fig. 3. Secuencia nucleotídica parcial del exón 14 del gen TFGBI en DNA de la paciente en la
que se demuestra una mutación puntual (flecha) de adenina (pico verde) a guanina (pico negro).
Debido a que la mutación es heterocígota, se observa la señal de los dos alelos en esa posición.
La mutación cambia un codón CAT a un codón CGT, lo que ocasiona que el aminoácido 626 de la
proteína TGFBI cambie de histidina a arginina (H626R).
Discusión
Las distrofias corneales son un grupo de alteraciones hereditarias caracterizadas por depósitos anormales de material amiloide, hialino o mixto en la córnea que ocasionan disminución importante del índice refractivo y pérdida de la transparencia corneales. En este estudio se realizó el análisis molecular del gen TGFBI en una familia mexicana afectada por distrofia corneal estromal granular tipo I (clásica). Estudios previos en sujetos de diversos orígenes étnicos han demostrado que este tipo de distrofia es causada casi exclusivamente por la sustitución de arginina a triptofano en el aminoácido 555 (R555W) de TGFBI. En la paciente analizada en este trabajo, el fenotipo corneal es compatible con una distrofia granular tipo I o clásica. El diagnóstico de distrofia granular tipo II o de Avellino, entidad que tiene algunas semejanzas clínicas con la distrofia granular tipo I, fue descartado debido a que en aquella se observan opacidades más gruesas, con morfología de anillos, discos o copos de nieve, que no fueron evidentes en nuestra paciente. Los familiares afectados no estuvieron disponibles para la valoración de las lesiones corneales y así poder establecer si existía variación clínica intrafamiliar. Sin embargo, en un hermano de la paciente que fue referido como afectado, el análisis molecular demostró que porta la misma mutación, confirmando así su estatus de afectado y el carácter familiar de la distrofia corneal.
La mutación identificada en la propósita y su hermano origina un cambio de histidina a arginina en el aminoácido 626 (H626R) de la proteína TGFBI. Éste es un hallazgo inesperado e interesante ya que esta mutación había sido identificada hasta ahora solamente en sujetos con la variante I/IIIA de distrofia corneal estromal en empalizada, una distrofia corneal fenotípicamente distinta a la de tipo granular (3,6,14). La biomicroscopía de la paciente no demostró lesiones lineales ramificadas típicas de las distrofias estromales de tipo empalizada. Esta es la primera demostración de que una distrofia granular resulta de la mutación H626R en TGFBI, dato que permite expandir el espectro de correlaciones fenotipo-molecular en este grupo de enfermedades corneales y hace evidente la variabilidad clínica asociada a una misma mutación en TGFBI. Previamente se han descrito ejemplos de excepciones a la aparente correlación genotipo-fenotipo estricta en la distrofia granular clásica, ya que en al menos dos sujetos no relacionados afectados con este tipo de distrofia se detectó una mutación de C a A en el nucleótido 417 de TGFBI, que lleva a la sustitución de arginina a serina en la posición 124 (Arg124Ser) (6,19).
Aunque en la mayoría de los casos de distrofias corneales asociadas a TGFBI existe un correlación estricta entre el genotipo y el fenotipo, se está haciendo evidente que en algunos casos, es posible que factores genéticos adicionales y/o ambientales sean capaces de modular la expresión fenotípica de mutaciones idénticas en este gen. La participación de tales genes «modificadores» ha sido sugerida para otras enfermedades monogénicas (20,21).
La caracterización de defectos moleculares específicos causantes de distrofias corneales en casos esporádicos o familiares permite identificar tempranamente a otros sujetos de la familia que porten el gen mutante y quienes desarrollarán la enfermedad. Esta información es de gran importancia no sólo en el asesoramiento genético familiar sino también en el posible diseño y aplicación de tratamientos futuros encaminados a retrasar o a inhibir el depósito del material que lleva finalmente al desarrollo de la distrofia corneal. El análisis de casos adicionales de distrofias corneales de distintas poblaciones permitirá establecer el espectro de mutaciones de TGFBI asociadas a este grupo de distrofias estromales y definir los fenotipos corneales que resultan de mutaciones específicas.
Agradecimientos
Los autores agradecen el apoyo del Patronato de la Fundación «Conde de Valenciana» para la realización de este trabajo.
Bibliografía
1. Chang SW, Tuli S, Azar DT. Corneal Dystrophies. In: Traboulsi EI. Genetic Diseases of the Eye. New York: Oxford University Press; 1998; 217-266. [ Links ]
2. Klintworth GK. The molecular genetics of the corneal dystrophies--current status. Front Biosci 2003; 8: 687-713. [ Links ]
3. Klintworth GK. Advances in the molecular genetics of corneal dystrophies. Am J Ophthalmol 1999; 128: 747-754. [ Links ]
4. Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 1997; 15: 247-251. [ Links ]
5. Korvatska E, Munier FL, Djemai A, Wang MX, Frueh B, Chiou AG, et al. Mutation hot spots in 5q31-linked corneal dystrophies. Am J Hum Genet 1998; 62: 320-324. [ Links ]
6. Munier FL, Frueh BE, Othenin-Girard P, Uffer S, Cousin P, Wang MX, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci 2002; 43: 949-954. [ Links ]
7. Ellies P, Renard G, Valleix S, Boelle PY, Dighiero P. Clinical outcome of eight BIGH3-linked corneal dystrophies. Ophthalmology 2002; 109: 793-797. [ Links ]
8. Afshari NA, Mullally JE, Afshari MA, Steinert RF, Adamis AP, Azar DT, et al. Survey of patients with granular, lattice, avellino, and Reis-Bucklers corneal dystrophies for mutations in the BIGH3 and gelsolin genes. Arch Ophthalmol 2001; 119: 16-22. [ Links ]
9. El-Ashry MF, El-Aziz MM, Larkin DF, Clarke B, Cree IA, Hardcastle AJ, et al. A clinical, histopathological, and genetic study of Avellino corneal dystrophy in British families. Br J Ophthalmol 2003; 87: 839-842. [ Links ]
10. Rozzo C, Fossarello M, Galleri G, Sole G, Serru A, Orzalesi N, et al. A common beta ig-h3 gene mutation (delta f540) in a large cohort of Sardinian Reis Bucklers corneal dystrophy patients. Mutations in brief no.180. Online. Hum Mutat 1998; 12: 215-216. [ Links ]
11. Okada M, Yamamoto S, Tsujikawa M, Watanabe H, Inoue Y, Maeda N, et al. Two distinct kerato-epithelin mutations in Reis-Bucklers corneal dystrophy. Am J Ophthalmol 1998; 126: 535-542. [ Links ]
12. Chakravarthi SV, Kannabiran C, Sridhar MS, Vemuganti GK. TGFBI gene mutations causing lattice and granular corneal dystrophies in Indian patients. Invest Ophthalmol Vis Sci 2005; 46: 121-125. [ Links ]
13. Aldave AJ, Gutmark JG, Yellore VS, Affeldt JA, Meallet MA, Udar N, et al. Lattice corneal dystrophy associated with the Ala546Asp and Pro551Gln missense changes in the TGFBI gene. Am J Ophthalmol 2004; 138: 772-781. [ Links ]
14. Chau HM, Ha NT, Cung LX, Thanh TK, Fujiki K, Murakami A, et al. H626R and R124C mutations of the TGFBI (BIGH3) gene caused lattice corneal dystrophy in Vietnamese people. Br J Ophthalmol 2003; 87: 686-689. [ Links ]
15. Korvatska E, Munier FL, Chaubert P, Wang MX, Mashima Y, Yamada M, et al. On the role of kerato-epithelin in the pathogenesis of 5q31-linked corneal dystrophies. Invest Ophthalmol Vis Sci 1999; 40: 2213-2219. [ Links ]
16. Collin HB, Hendicott PL. Granular dystrophy of the cornea. Clin Exp Optom 1999; 82: 203-206. [ Links ]
17. Moller HU. Granular corneal dystrophy Groenouw type I: clinical and genetic aspects. Acta Ophthalmol Suppl 1991; 198: 1-40. [ Links ]
18. Haddad R, Font RL, Fine BS. Unusual superficial variant of granular dystrophy of the cornea. Am J Ophthalmol 1977; 83: 213-218. [ Links ]
19. Stewart HS, Ridgway AE, Dixon MJ, Bonshek R, Parveen R, Black G. Heterogeneity in granular corneal dystrophy: identification of three causative mutations in the TGFBI (BIGH3) gene-lessons for corneal amyloidogenesis. Hum Mutat 1999; 14: 126-132. [ Links ]
20. Nadeau JH. Modifier genes and protective alleles in humans and mice. Curr Opin Genet Dev 2003; 13: 290-295. [ Links ]
21. Haider NB, Ikeda A, Naggert JK, Nishina PM. Genetic modifiers of vision and hearing. Hum Mol Genet 2002; 11: 1195-1206. [ Links ]
![]() Dirección para correspondencia
Dirección para correspondencia
Juan Carlos Zenteno
Unidad de Investigación, Instituto de Oftalmología Conde de Valenciana
Chimalpopoca 14, Col. Obrera
México D.F. 06800
E-mail: jczenteno@salud.gob.mx
Recibido: 20.10.05.
Aceptado: 19.7.06.

