










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versão impressa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.83 no.3 Mar. 2008
Detección de deleciones en DNA mitocondrial heteroplásmico por medio de PCR en el Síndrome de Kearns-Sayre
PCR-based detection of heteroplasmic deleted mitochondrial DNA in Kearns-Sayre Syndrome
Ramírez-Miranda A.1, Navas-Pérez A.2, Gurria-Quintana L.2, Vargas-Ortega J.2, Murillo-Correa C.2, Zenteno J.C.1,3
Instituto de Oftalmología Conde de Valenciana. México DF. México.
1 Doctor en Medicina. Departamento de Genética.
2 Doctor en Medicina. Departamento de Estrabismo.
3 Doctor en Ciencias. Unidad de Investigación.
Dirección para correspondencia
RESUMEN
Objetivo: El síndrome de Kearns-Sayre (SKS) es un trastorno neuromuscular causado por defectos genéticos en el DNA mitocondrial siendo deleciones de tamaño variable la alteración mas común. Se describen las características clínicas y los resultados del análisis molecular del DNA mitocondrial en una paciente Mexicana con Síndrome de Kearns-Sayre.
Métodos: Examen oftalmológico completo, caracterización fenotípica del Síndrome de Kearns-Sayre y análisis del DNA mitocondrial mediante reacción en cadena de la polimerasa (PCR). Se discuten correlaciones genotipo-fenotipo.
Resultados: La paciente afectada mostró ptosis, oftalmoplejia progresiva externa cambios pigmentarios en retina periférica y bloqueo de rama derecha del haz de His. Los análisis moleculares revelaron una deleción de ~5 kb en el DNA mitocondrial además de trazas de DNA sin alteraciones indicando heteroplasmia.
Conclusiones: El análisis molecular del DNA mitocondrial confirmó el diagnostico clínico de síndrome de Kearns-Sayre. La utilización de PCR, como se describe en este trabajo, es un método rápido y económico para el diagnóstico de la deleción común de 4977 pb en el síndrome de Kearns-Sayre. En estos casos, el diagnóstico por PCR evitaría procedimientos diagnósticos invasivos y traumáticos como biopsia muscular o punción lumbar.
Palabras Clave: Síndrome de Kearns-Sayre; estrabismo; oftalmoplejia; DNA mitocondrial; heteroplasmia.
ABSTRACT
Objective: To describe the clinical data and the results of molecular analyses of the mitochondrial DNA in a patient with Kearns-Sayre Syndrome.
Methods: Molecular analyses of mitochondrial DNA from the patient included PCR amplification of a region where the common Kearns- Sayre deletion is located and Genotype-Phenotype correlations are discussed.
Results: The affected patient showed ptosis, progressive external ophthalmoplegia, pigmentary changes in the peripheral retina and right bundle block. Molecular analysis disclosed a ~5kb deletion in the mitochondrial DNA and some wild type mtDNA indicating heteroplasmy.
Conclusions: Molecular analysis of mitochondrial DNA confirmed the clinical diagnosis of Kearns-Sayre syndrome. PCR provides a rapid method to identify the common 4997 bp deletion in Kearns-Sayre syndrome. In such cases, PCR diagnosis could avoid invasive methods such as muscle biopsy or spinal tap (Arch Soc Esp Oftalmol 2008; 83: 155-160).
Key words: Kearns-Sayre syndrome, strabismus, ophthalmoplegia, mitochondrial DNA, heteroplasmy.
Introducción
Las citopatías mitocondriales son un grupo poco común de enfermedades multisistémicas en las que existe evidencia bioquímica, histopatológica y/o genética de disfunción mitocondrial (1). Estas enfermedades se manifiestan como síndromes clínicos muy bien definidos y son causadas por defectos moleculares en el DNA mitocondrial (mtDNA) que pueden variar desde mutaciones puntuales a rearreglos génicos mayores como duplicaciones o deleciones (2). Los tejidos con gran demanda metabólica de energía oxidativa, tales como el músculo y el cerebro son particularmente susceptibles a las mutaciones del mtDNA a pesar de que las enfermedades relacionadas al mtDNA pueden ser diferenciadas en base a sus características clínicas, la mayoría de ellas comparten las manifestaciones de acidosis láctica y proliferación masiva de mitocondrias en el músculo, que origina un patrón histopatológico típico de fibras rojas-rasgadas (3). Las manifestaciones clínicas se inician generalmente en la infancia y pueden incluir acidosis láctica, anemia, miopatía, anormalidades neurológicas, alteraciones endocrinas, enfermedades renales, sordera neurosensorial, distrofia retiniana atípica, y defectos en el sistema de conducción cardiaca, siendo estos últimos la principal causa de muerte prematura.
Las tres principales enfermedades asociadas a deleciones del mtDNA son el síndrome de Pearson, la oftalmoplejia externa progresiva crónica y el síndrome de Kearns-Sayre (SKS, OMIM #530000) (4). El SKS es un trastorno pleiotrópico, descrito por primera vez en 1958 y que se caracteriza principalmente por oftalmoplejia externa progresiva crónica, retinopatía pigmentaria, bloqueo cardiaco y ataxia cerebelosa (5).
La edad usual para su inicio es antes de los 20 años de edad y los sujetos afectados frecuentemente presentan datos clínicos asociados como sordera y alta concentración de proteínas en el líquido cefalorraquídeo (6). Son también comunes síntomas endocrinos tales como diabetes, hiperaldosteronismo y amenorrea.
Las deleciones en el mtDNA asociadas al SKS son variables y fluctúan desde 1,3 hasta 8 kilobases (kb) con distintas cantidades de mtDNA delecionado presentes en diferentes tejidos, lo que se correlaciona con la naturaleza multisistémica del síndrome (7-9). El rearreglo mas frecuente observado en el mtDNA de pacientes con SKS es la «deleción común» de 4977pb (mtDNA4977), localizada entre los nucleótidos 8482 y 13640 (10) del mtDNA y que se encuentra en aproximadamente 50% de los sujetos afectados (10). Además de las deleciones del mtDNA mencionadas, se han identificado también mutaciones puntuales en algunos pacientes de SKS (6).
Existen pocas publicaciones que describan una investigación oftalmológica completa de pacientes con SKS así como análisis molecular del mtDNA. En este trabajo se presentan los resultados de una evaluación oftalmológica completa así como el análisis del mtDNA en una paciente mexicana con diagnóstico de SKS mediante técnica de PCR que permite identificar la deleción común en el mtDNA de los pacientes con SKS tanto en estado de heteroplasmia como de homoplasmia.
Sujetos, material y métodos
Una paciente de 15 años de edad fue referida a nuestro hospital debido a ptosis y limitación a los movimientos oculares bilaterales de 2 años de evolución. La paciente no contaba con antecedentes de diabetes, hipertensión o trauma, contaba con el antecedente ginecológico de menarquia a los 13 años con ciclos regulares y no se encontraron antecedentes familiares de ptosis, miopatías o enfermedades hereditarias. A la exploración física la estatura de la paciente fue de 160 cm, y la exploración neurológica no mostraba evidencia de distrofia muscular, ataxia, o sordera. A la exploración oftalmológica, la agudeza visual en el ojo derecho (OD) fue de 20/40 y en el izquierdo (OI) de 20/25; las presiones intraoculares fueron de 11 mmHg (OD) y 12 mmHg (OI). La exploración de movimientos oculares mostró marcada limitación en todas las direcciones de la mirada para ambos ojos, con una exotropia de 15 dioptrías prismáticas (fig. 1). La apertura palpebral era de 7 mm en ambos ojos, con función muy disminuida del elevador en ambos ojos (5 mm) y reflejo pupila margen de 1 mm en ambos ojos. El diámetro pupilar se encontraba dentro del rango normal y los reflejos pupilares se hallaban intactos. No se encontraron alteraciones en el segmento anterior de ambos ojos. La exploración bajo dilatación pupilar del fondo de ojo reveló múltiples cambios pigmentarios con un patrón en «sal y pimienta» en el ecuador y la periferia retiniana de ambos ojos (fig. 2). La angiografía con fluoresceína mostró atrofia moderada del epitelio pigmentado de la retina (fig. 3), mientras que la electroretinografía evidenció disminución del umbral a respuesta de estímulos luminosos, previa dilatación y adaptación a la oscuridad. La electromiografía cinetológica demostró afección de todos los músculos extraoculares, del músculo frontal y de músculos faríngeos. En la electrocardiografía de 12 derivaciones se encontró bloqueo incompleto de rama derecha del haz de His.
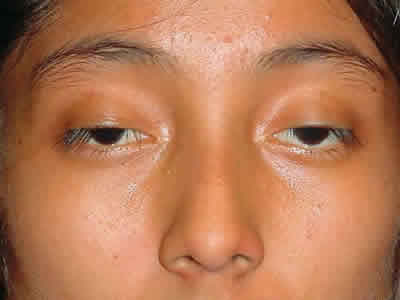
Fig. 1. Ptosis bilateral y oftalmoplejía en una paciente de
15 años de edad con el diagnóstico de Síndrome de Kearns-Sayre.

Fig. 2. Apariencia del polo posterior mostrando cambios
pigmentarios y palidez de cabeza de nervio óptico.

Fig. 3. Angiografía con fluoresceína mostrando atrofia difusa
del epitelio pigmentado de la retina.
El análisis citológico y químico del líquido cefalorraquídeo obtenido por punción lumbar mostró niveles elevados de proteínas (124 mg/dl). La paciente fue diagnosticada clínicamente de Síndrome de Kearns-Sayre y fue referida al departamento de Genética.
Estudios Moleculares del DNA mitocondrial: La investigación se realizó previa autorización por el comité de Bioética de la Institución y firma de consentimiento informado por los padres de la paciente. A partir de una muestra de 8 ml de sangre periférica se obtuvo un concentrado plaquetario del que se aisló el mtDNA con la ayuda del QIAamp DNA mini kit (Qiagen, Hilden, Alemania), siguiendo las indicaciones del fabricante. Se amplificó el mtDNA por medio de PCR utilizando los cebadores MT-1 y MT-4 (indicados en la tabla I), tomados de Brenner et al (13). MT-1 flanquea la región del mtDNA de la «deleción común» del SKS en la dirección 5, mientras que MT-4 flanquea el sitio de la deleción en sentido 3. Este par de cebadores amplifica un producto de ~5kb: si la «deleción común» de 4977pb del SKS, está presente, el producto de PCR obtenido será de 550 pb; en caso de que la deleción esté ausente, el producto esperado de ~5kb será amplificado; el mtDNA heteroplásmico mostrará ambas bandas. Cada 25 µl de reacción de PCR incluyeron 150 ngs de mtDNA, MgCl2 a una concentración final de 1,5 mM, 200 µM de cada dNTP, 1 µM de cada cebador, y 1,25 U de HotStart Taq polimerasa (Quiagen). Los cebadores MT-1 y MT-4 fueron utilizados y el programa de temperaturas incluyó un ciclo inicial de desnaturalización a 95ºC por 15 minutos, 40 ciclos de desnaturalización a 95ºC por un minuto, alineamiento a 65,2ºC por minuto y extensión a 72°C por 3 minutos; con un ciclo de extensión final a 72ºC por 10 minutos.

Se utilizó un marcador estándar de DNA de 100 pb para confirmar el tamaño de los productos amplificados. Para descartar la posibilidad de contaminación causada por la amplificación mediante PCR, en cada reacción de PCR se incluyó un tubo en el cual el templado de DNA fue omitido y sustituido por H2O el cual sirvió como control negativo.
Resultados
La Amplificación mediante PCR del mtDNA de la paciente con SKS mostró la presencia de dos productos: una banda de 550 pb que indica la presencia de la «deleción común» de ~5 kb y una banda de mtDNA silvestre (fig. 4, línea 3), lo cual demuestra la presencia de mitocondrias con DNA normal y otras con DNA con la deleción (heteroplasmia); en contraste, la amplificación por PCR en el mtDNA de un sujeto normal evidenció solo una banda de 5 kb excluyendo la presencia de deleción (fig. 4, línea 2). Estos resultados fueron consistentes después de 3 amplificaciones independientes de mtDNA de la paciente y control positivo. En ningún caso, el control negativo produjo amplificación del mtDNA, excluyendo amplificación debida a contaminación por DNA.

Fig. 4. Productos amplificados de mtDNA por medio de PCR.
Línea 2: DNA control mostrando una banda de alto peso molecular
correspondiente a mtDNA normal de ~5 kb; línea 3: DNA de la
paciente mostrando una banda de 550 pb indicativa de la deleción
común y una banda menos intensa de ~5 kb correspondiente
al mtDNA silvestre, demostrando heteroplasmia mitocondrial.
Discusión
El SKS es una enfermedad neuromuscular poco frecuente caracterizada por un inicio antes de los 20 años de edad, oftalmoplejia externa progresiva crónica, y degeneración pigmentaria de la retina. Otros hallazgos frecuentes en los individuos afectados incluyen defectos en el sistema de conducción cardiaca, ataxia cerebelosa y concentración elevada de proteínas en liquido cefalorraquídeo (>100 mg/dl).
Los pacientes con SKS presentan ptosis bilateral como uno de los primeros signos de la enfermedad seguida por oftalmoplejia externa progresiva crónica algunos años después. En nuestro caso tanto las lateroversiones como la supra e infraversión se encontraron afectadas, indicando una forma severa de la enfermedad; los defectos en el sistema de conducción cardiaca son comunes en el síndrome (se presentan en 57% de los pacientes) y en nuestro caso se documentó electrocardiográficamente solo un bloqueo incompleto leve; estos datos nos indican que existe heterogeneidad fenotípica en el SKS, un hecho que puede generar retos y dificultades en el diagnóstico de este síndrome en algunos pacientes. En este contexto es interesante destacar que la «deleción común» del SKS en el mtDNA se encuentra fuertemente relacionada con las alteraciones de conducción cardiaca (8,15). Nuestro caso demuestra y confirma esta correlación. El seguimiento cardiovascular está indicado en todo paciente con SKS, para así identificar y tratar tempranamente las alteraciones y complicaciones graves de aparición tardía.
En el SKS, así como en otros trastornos relacionados con el mtDNA, puede encontrarse heteroplasmia, una mezcla de mtDNA mutante con silvestre en la misma célula o tejido. La relación de DNA mutante con DNA silvestre es de suma importancia para determinar la severidad del fenotipo en un desorden mitocondrial. La ausencia de ataxia cerebelosa y sordera neurosensorial en nuestra paciente podría deberse a este fenómeno. Por otro lado, la biopsia de músculo estriado es uno de lo métodos usuales para el diagnóstico histopatológico de SKS, siendo las fibras rojas-rasgadas el hallazgo característico en el músculo afectado.
Sin embargo, el desarrollo de procedimientos diagnósticos no invasivos como la demostración de la «deleción común» de SKS por medio de PCR, puede evitar la necesidad de toma de biopsias, facilitando el abordaje de los pacientes con sospecha de SKS. La PCR es una útil alternativa para los casos en los que el paciente rechaza la biopsia muscular como método diagnóstico.
Los métodos por PCR para detectar la «deleción común» de SKS están basados en el hecho de que si la deleción está ausente el fragmento de 5 kb no será visualizado por su gran tamaño para amplificación; contrariamente, si la deleción esta presente, el pequeño fragmento remanente de mtDNA, será fácilmente amplificado y detectado (10,13). Una clara desventaja de este abordaje es la imposibilidad de detectar heteroplasmia. En el método descrito en este trabajo fue posible amplificar el pequeño fragmento indicativo de la «deleción común» de SKS así como el fragmento de ~5 kb de mtDNA silvestre, demostrando así la presencia de mitocondrias heteroplásmicas. Similarmente, este método podría detectar homoplasmia para mtDNA mutante o normal del tejido investigado.
Agradecimientos
Los Autores agradecen al patronato de la Fundación «Conde de Valenciana» por el apoyo para realizar este trabajo.
Bibliografía
1. Leonard JV, Schapira AH. Mitochondrial respiratory chain disorders. I: mitochondrial DNA defects. Lancet 2000; 355: 299-304. [ Links ]
2. Johns DR. Seminars in Medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N Engl J Med 1995; 333: 638-644. [ Links ]
3. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003; 348: 2656-2668. [ Links ]
4. Blakely EL, He L, Taylor RW, Chinnery P F, Lightowlers R N, Schaefer A M, et al. Mitochondrial DNA deletion in «identical» twins brothers. J Med Genet 2004; 41: e19. [ Links ]
5. Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmoplegia and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol 1958; 60: 280-289. [ Links ]
6. Degoul F, Nelson I, Lestienne P, Francois D, Romero N, Duboc D. Deletions of mitochondrial DNA in Kearns-Sayre syndrome and ocular myopathies: genetic, biochemical and morphological studies. J Neurol Sci 1991; 101: 168-177. [ Links ]
7. Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, Schon EA.. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988; 38: 1339-1346. [ Links ]
8. Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. New Eng J Med 1989; 320: 1293-1299. [ Links ]
9. Larsson NG, Holme E, Kristiansson B, Oldfors A, Tulinius M. Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediat Res 1990; 28: 131-136. [ Links ]
10. Barritt JA, Brenner CA, Cohen J, Matt DW. Mitochondrial DNA rearrangements in human oocytes and embryos. Mol Hum Reprod 1999; 5: 927-933. [ Links ]
11. Seneca S, Verhelst H, De Meirleir L, Meire F, Ceuterick-De Groote C. A new mitochondrial point mutation in the transfer RNA(Leu) gene in a patient with a clinical phenotype resembling Kearns-Sayre syndrome. Arch Neurol 2001; 58: 1113-1118. [ Links ]
12. Bosbach S, Kornblum C, Schroder R, Wagnere M. Executive and visuospatial deficits in patients with chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome. Brain 2003; 126: 1231-1240. [ Links ]
13. Brenner CA, Wolny YM, Barrit JA, Matt DW, Munne S, Cohen J. Mitochondrial DNA deletion in human oocytes and embryos. Mol Hum Reprod 1998; 4: 887-892. [ Links ]
14. Simaan EM, Mikati MA, Touma EH, Ritig A. Unusual presentation of Kearns-Sayre syndrome in early childhood. Pediatr Neurol 1999; 21: 830-831. [ Links ]
15. Young TJ, Shah AK, Lee MH, Hayes DL. Kearns-Sayre syndrome: a case report and review of cardiovascular complications. Pacing Clin Electrophysiol 2005; 28: 454-457. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Arturo Ramírez-Miranda
Chimalpopoca 14 Col. Obrera 06800
México DF, México
E-mail: arturorammir@gmail.com
Recibido: 7/11/06.
Aceptado: 4/2/08.

