










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
Print version ISSN 0365-6691
Arch Soc Esp Oftalmol vol.86 n.10 Oct. 2011
Retinosquisis ligada al cromosoma X, presentación inusual: estrabismo
X linked retinoschisis, unusual presentation: strabismus
A.B. Areizaga Osésa, R. Martínez Fernándezb, M. Galdos Iztuetab y N. Muruzabal Zaldíbarc
aUnidad de Oftalmología, Hospital San Eloy, Baracaldo, Vizcaya, España
bSección Oftalmología pediátrica, Hospital Cruces, Baracaldo, Vizcaya, España
cUnidad de Oftalmología, Hospital Cruces, Baracaldo, Vizcaya, España
Dirección para correspondencia
RESUMEN
Caso clínico: La retinosquisis ligada al cromosoma X es una retinopatía degenerativa de carácter recesivo. Presentamos dos casos clínicos que debutaron con una presentación atípica (estrabismo) en la infancia precoz (lactancia). Ambos niños presentaban velos vítreos en retina periférica. Se encontró una mutación en el gen XLRS1 en ambos casos.
Discusión: La retinosquisis ligada al cromosoma X es una de las causas principales de degeneración macular en niños varones. Se caracteriza por una esquisis foveal estrellada, asociada o no a retinosquisis periférica. El diagnóstico clínico puede ser difícil por la alta variabilidad fenotípica del cuadro. Además, el ERG y la OCT pueden ser normales en fases precoces de la enfermedad y de difícil realización en niños pequeños. Consideramos que el cribado para la mutación del gen XLRS1 es útil para evaluar casos con presentación atípica de retinosquisis ligada al cromosoma X y/o en niños en los que otras pruebas complementarias no son realizables.
Palabras clave: Retinopatía. Retinosquisis. Estrabismo.
ABSTRACT
Case report: X linked retinoschisis is a recessively inherited degenerative retinopathy. We report two cases that debuted with an unusual presentation (strabismus) in early childhood (months). Both of them presented with vitreous veils in the retinal periphery. Mutation in the XLRS1 gene was detected in both cases.
Discussion: X linked retinoschisis is one of the leading causes of macular degeneration in male children. Clinical features include a stellate foveal schisis, with or without peripheral retinoschisis. Clinical diagnosis is often difficult because of a high degree of phenotype variability. Furthermore, ERG and OCT may be normal in early stages of the disease. In our opinion, the XLRS1 gene mutation screening provides a powerful clinical tool for evaluating clinically ambiguous cases of X linked retinoschisis.
Key words: Retinopathy. Retinoschisis. Strabismus.
Introducción
La retinosquisis ligada al cromosoma X descrita por Hass en 1898 como una pérdida visual precoz asociada a esquisis foveal bilateral1, es la causa más frecuente de degeneración macular en niños.
La prevalencia de la enfermedad es baja: 1/120.000 (excepto en Finlandia: 1/20-000), por mutaciones en poblaciones fundadoras del país)1. Por tratarse de herencia recesiva ligada al cromosoma X, se presenta casi exclusivamente en varones. Aunque se han descrito casos de mujeres afectadas con antecedentes de consanguinidad2.
La forma más frecuente de presentación es una disminución de la agudeza visual. Es típico el debut con dificultad para la lectura en edad escolar (5-10 años). La esquisis foveal bilateral es el hallazgo clínico más frecuente (68-100% de los casos)1. El 50% de los pacientes presentan una esquisis periférica, típicamente en retina inferotemporal. Esta esquisis suele ser muy llamativa en la infancia, cuando puede producir nistagmus o estrabismo debido a su gran tamaño, e incluso puede afectar al campo visual1. Típicamente se produce una remisión espontánea de la esquisis, dejando una línea de demarcación cuando se resuelve.
Casos clínicos
Caso 1
Lactante varón de 6 meses (afecto) y en heterocigosis en su madreal que atendimos en nuestra consulta por presentar estrabismo divergente con ausencia de fijación en ojo izquierdo.
Tanto el embarazo como el parto habían transcurrido sin complicaciones. Como antecedente personal mencionar que el lactante había sufridoun accidente de tráfico el mes previo.
A la exploración el polo anterior era normal en ambos ojos.
En el fondo de ojo izquierdo se observó un desprendimiento de retina inferior asociado a un velo vítreo con tracción retiniana hasta la papila. (Fig. 1)
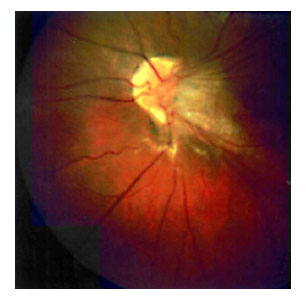
Fig. 1. Tracción retiniana hasta la papila
en ojo izquierdo.
En el fondo de ojo derecho no se observaron alteraciones retinianas.
En la retinoscopia presentó un punto neutro de +3.00 -2.00 a 10o en el ojo derecho y +3.50 +0.50 a 7o en el ojo izquierdo.
Dados los antecedentes del accidente de tráfico, se atribuyó un origen traumático al desprendimiento retiniano en el ojo izquierdo.
Se realizaron controles periódicos. El desprendimiento de retina evolucionó hacia la reabsorción completa, apareciendo una línea de demarcación en retina temporal. La mácula se adaptó, presentando una alteración a nivel del epitelio pigmentario de la retina. (Fig. 2)

Fig. 2. Esquisis periférica temporal junto
con línea de demarcación en ojo derecho.
A los meses se observaron bridas vítreas en arcada temporal inferior en el ojo derecho. Esto condujo a pensar en una posible retinosquisis periférica como origen del desprendimiento de retina del ojo izquierdo.
Se realizaron tanto OCT macular como ERG, sin que los resultados fueran valorables en ambas pruebas por la mala colaboración del paciente dada su corta edad. Por ello se realizó estudio de ADN a partir de una muestra de sangre periférica. Se observó una mutación (Q154R) en el exón 5 del gen XLRS responsable de la retinosquisis ligada al cromosoma X. La mutación se presentaba en hemicigosis en el paciente (afecto) y en heterocigosis en su madre (portadora).
A la edad de 6 años, la agudeza visual era de 20/20 en ojo derecho y 20/400 en ojo izquierdo, a pesar de tratamiento prolongado con oclusiones horarias del ojo derecho.
En la exploración se observó una condensación vítrea temporal inferior en el ojo derecho y una retinosquisis periférica temporal inferior en el ojo izquierdo, asociada a fibrosis residual de la arcada temporal inferior. (Fig. 3)
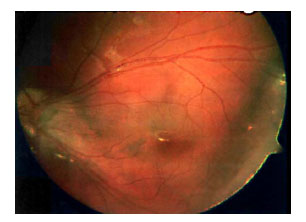
Fig. 3. Fibrosis residual de la arcada
temporal inferior en ojo izquierdo.
Destacó la ausencia de esquisis foveal mediante OCT en ambos ojos.
Caso 2
Paciente varón de 11 meses al que atendimos en nuestra consulta para estudio de estrabismo convergente con ausencia de fijación por ojo derecho.
A la exploración presentaba un punto neutro de +5.50 -1.00 a 10o en ojo derecho y +8.00 -1.00 a 10o en ojo izquierdo. Una endotropia de +10o, dominancia de OI.
El polo anterior no presentaba alteraciones en ambos ojos.
En el fondo de ojo derecho no se apreciaron alteraciones significativas, salvo un velo vítreo periférico temporal inferior. (Fig. 4)
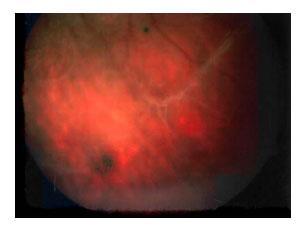
Fig. 4. Velo vítreo periférico temporal
inferior en ojo derecho.
El fondo de ojo en el ojo izquierdo era normal.
Se inició tratamiento con oclusión de 4 h/día en ojo izquierdo. En revisiones periódicas, se observó una condensación vítrea a nivel temporal inferior, sin tracciones ni desprendimiento de retina en el ojo derecho.
Ante la falta de respuesta al tratamiento con oclusiones, se realizó ERG y PEV, con resultados dentro de la normalidad.
En la evolución se apreció una esquisis retiniana periférica bajo la condensación vítrea en ojo derecho. (Fig. 5)
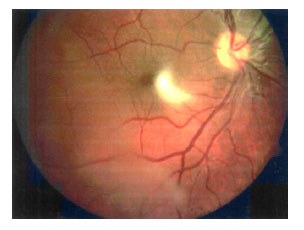
Fig. 5. Esquisis periférica temporal
inferior en ojo izquierdo.
Se realizó estudio genético mediante muestra de sangre periférica. Se observó una mutación en el exón 5 del gen RS1 localizado en el cromosoma Xp22.2-p22.1.
El paciente fue diagnosticado de retinosquisis ligada al cromosoma X y su madre de portadora.
A la edad de 5 años, presentaba una agudeza visual de 20/125 en el ojo derecho y 20/40 en el ojo izquierdo, a pesar de tratamiento con oclusiones.
En el fondo de ojo presentaba una esquisis retiniana temporal inferior en ojo derecho, alcanzando polo posterior, sin afectación macular y una esquisis periférica temporal en ojo izquierdo.
No se apreciaron alteraciones maculares en OCT en ambos ojos. (Fig. 6)
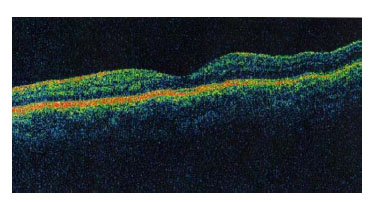
Fig. 6. OCT sin alteraciones en ojo izquierdo.
Discusión
El diagnóstico clínico de la retinosquisis ligada al cromosoma X puede ser difícil por el alto grado de variabilidad fenotípica3.
Clásicamente el diagnóstico se basaba en el carácter hereditario ligado al cromosoma X y la negatividad de la onda b en el ERG escotópico. Actualmente, la OCT es un método útil para estudiar los quistes a nivel foveal3. En el Oxford Eye Hospital se ha desarrollado un método para realizar OCT, incluso de retina periférica, a neonatos, bajo anestesia general4.
El ERG puede ser normal en fases precoces de la enfermedad, o bien puede estar alterado por la presencia de alteraciones en la retina3,5.
La confirmación genética es el diagnóstico definitivo en casos con clínica atípica y ausencia de familiares afectos3.
El gen de la retinosquisis ligada al X (XLRS1) se encuentra en el brazo corto distal del cromosoma X(Xp22)2. Este gen codifica la proteína retinosquisina, secretada por fotorreceptores y células bipolares. Se trata de una proteína implicada en la diferenciación retiniana temprana, así como en la adhesividad y señalización intercelular7. Secundariamente se produce una alteración de las células de Müller, por el acúmulo de esta retinosquisina mutada.
Esto explicaría los hallazgos observados en estudios con OCT4,5, donde se observa la esquisis inicial a nivel de la capa nuclear interna (cuerpos de células de Müller y células bipolares). Por otro lado, al estar dañadas las células de Müller, responsables del control del K extracelular, se explicaría la alteración típica del ERG6.
El pronóstico visual de la retinosquisis ligada al X es discutido. Se considera que la agudeza visual se mantiene estable tras un empeoramiento marcado a la edad escolar, produciéndose un empeoramiento posterior a partir de los 40-50 años por atrofia macular.
En cuanto al tratamiento, este va dirigido a controlar las posibles complicaciones (DR, hemorragia vítrea, ambliopía...,). No está indicado el tratamiento profiláctico con láser alrededor de las cavidades quísticas periféricas por las posibles complicaciones2.
Recientes estudios promulgan la vitrectomia, puesto que la tracción vítrea podría desempeñar un papel crucial en la retinosquisis (debido a la falta de adhesión celular coexistente). De este modo, eliminar el vítreo evitaría complicaciones como la hemorragia vítrea, atrofia macular, desprendimiento de retina...6.
Hemos presentado dos casos en los que la enfermedad debuta con estrabismo en la lactancia con la característica común de velos vítreos. Ante estos casos en los que generalmente las pruebas complementarias habituales son poco fiables, creemos que el diagnóstico genético es fundamental.
En el caso 2, estudios genéticos al resto de los familiares han dado como resultado el diagnóstico de la misma enfermedad a un hermano del paciente, aunque no presenta signos de la enfermedad. Se trata de una herencia ligada al cromosoma X, con penetrancia completa de la mutación, con una expresividad muy variable, incluso en miembros de la misma familia, de manera que la mutación puede encontrarse en varones de la familia, aunque no presenten clínica.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Tantri Avinash, Vrabec Tamara R, Cu-Unjieng Andrew, Frost Arcilee, Annesley William H, Donoso Larry A. X-Linked Retinoschisis: A Clinical and Molecular Genetic Review. Surv Ophtahlmol. 2004; 49:214-30. [ Links ]
2. Edwards Albert O, Joseph E, Robertson JR. Hereditary vitreoretinal degenerations. En: Stephen JRyan, eds. Retina. I, 4th ed N York: Mosby; 2006. p. 474-6. [ Links ]
3. Shukla D, rajendran A, Gibbs D, Suganthallakshmi B, Zhang K, Sundaresan P. Unusual manifestations of X-linked retinoschisis: clinical profile and diagnostic evaluation. Am J Ophthalmol. 2007; 144:419-23. [ Links ]
4. Dhingra S, Patel CK. Diagnosis and pathogenesis of congenital X-linked retinoschisis with optical coherence tomography. J Pediatric Ophthalmol Strabismus. 2010; 47:105-7. [ Links ]
5. Prenner JL, Capone A, Ciaccia S, Takada Y, Sieving PA, Trese MT. Congenital X-Linked Retinoschisis classification system. Retina. 2006; 26:S61-4. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Correo electrónico: aneareizaga@yahoo.es
(A. B. Areizaga Osés)
Recibido el 5 de julio de 2010
Aceptado el 25 de marzo de 2011

