










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkCirugía Plástica Ibero-Latinoamericana
versión On-line ISSN 1989-2055versión impresa ISSN 0376-7892
Cir. plást. iberolatinoam. vol.41 no.2 Madrid abr./jun. 2015
https://dx.doi.org/10.4321/S0376-78922015000200010
Primer caso clínico de Síndrome de Pai en México
First clinical case of Pai Syndrome in México
Melloni-Magnelli, L.F.*, de la Garza-Giacomán, R.**, Martínez-Leija, H.***, Guzmán-Rodríguez, R.****
* Especialista en Cirugía Plástica, Estética y Reconstructiva.
** Especialista en Otorrinolaringología.
*** Especialista en Radiología.
**** Licenciado en Enfermería especializado en Cirugía.
Hospital Regional Materno Infantil de Alta Especialidad de Monterrey, Monterrey, Nuevo León, México.
Dirección para correspondencia
RESUMEN
Descrito por primera vez en 1987, el Síndrome de Pai se considera una variante rara de la displasia fronto-nasal. Consiste en el fallo del cierre de la línea media y sus signos son encéfalo-cráneo-faciales. Los hallazgos que conforman el síndrome son: la presencia de una variedad de pólipos (intranasal, cutáneos y alveolar del maxilar superior), fisura ósea y labiopalatina en la línea media, lipoma intracraneal y agenesia parcial o total del cuerpo calloso.
Su causa es desconocida y su presentación esporádica. La incidencia se estima en 1 de cada 20.000 a 40.000 recién nacidos, siendo el sexo femenino el más afectado.
El objetivo de este artículo es presentar el primer caso clínico documentado en México con estas características, y de acuerdo al último caso publicado en 2014 por Mee Hong, es el número 38 de la literatura mundial.
Palabras clave: Síndrome de Pai, Pólipos nasales, Lipoma intracraneal, Agenesia de cuerpo calloso, Fisura media labiopalatina.
Nivel de evidencia científica: 5.
ABSTRACT
Described by the first time in 1987, Pai's Syndrome is considered a rare variant of the displasia fronto-nasal. It consists of the fault of the closing of the middle line and his signs are encephalo-craneo-facial. The findings of the syndrome are the presence of a variety of polyps (intranasal, cutaneous and alveolar of the upper jaw), bony and lip-palate cleft in the middle line, intracranial lipoma and partial or total agenesia of the corpus callosum.
The etiology of this syndrome is not known, and its presentation is sporadic. The incidence is estimated in 1 of every 20.000-40.000 newborn children, being the most affected feminine sex.
The aim of this article is to present the first clinical case reported in Mexico with these characteristics and that in agreement to the last case published in 2014 for Mee Hong, it is number 38 of the world literature.
Key words: Pay´s Syndrome, Nasal polyps, Intracraneal lipona, Agenesia of the corpus callosum, Median lip and cleft palate.
Level of evidence: 5.
Introducción
Definido por primera vez en 1987 por Pai (1), este síndrome consiste en el fallo del cierre de la línea media y sus signos son encéfalo-cráneo-faciales. Se caracteriza por la presencia de una variedad de pólipos (intranasal, cutáneos y alveolar del maxilar superior) todos ellos localizados en la línea centrofacial (2).
Los pólipos son el marcador principal ya que están presentes en el 100% de los casos publicados (1-16). Histológicamente se describen como tejido dermoide o hamar tomatoso; una combinación de piel, glándulas pilosebaceas y tejido graso normal (3,6,8). Son de diferentes tamaños y varían en número de 1 a 3 en cada paciente, aunque pudieran no estar presentes, y se localizan sobre el labio superior, en el dorso nasal e incluso en la frente. El pólipo congénito nasal emerge del septum nasal y por lo general es unilateral. El pólipo alveolar se localiza en la hendidura media superior, entre dos frenillos, con o sin labio hendido y hendiduras óseas del maxilar superior, que pueden ir desde una simple muesca hasta una fisura completa del labio y del paladar en la línea media (2-4,10).
Los lipomas intracraneales forman parte también de los signos del Síndrome de Pai. (3,6,11) y se localizan principalmente en la cisura interhemisférica. Los lipomas del cuerpo calloso presentan diversos grados de disgenesia llegando hasta la agenesia del cuerpo calloso. Actualmente se sabe que la formación de lipomas resulta de una persistencia anormal y una mala diferenciación de la meninge primitiva, un derivado mesenquimatoso de la cresta neural que normalmente da lugar a la formación de las cisternas subaracnoideas. Clínicamente, el 50% son asintomáticos o se asocian a síntomas neurológicos inespecíficos como retraso psicomotor, cefalea y epilepsia. Castori en el 2007 (8) recogió el primer caso de Síndrome de Pai con agenesia del cuerpo calloso, a diferencia de otras publicaciones previas en las que solo se mencionaba la presencia del lipoma intracraneal.
El Síndrome de Pai puede diagnosticarse mediante ultrasonido de control prenatal, como describen Chousta y col. (11) Andreeva y col. (12) y Ocak y col. (13). En el caso recogido por Blouet (14), y debido a los hallazgos del ultrasonido, los padres solicitaron terminar con el embarazo a las 35 semanas acogiéndose a la ley francesa por encontrar agenesia del cuerpo calloso, lipoma intracraneal de tipo túbulo-nodular, 2 pólipos cutáneos, nariz bífida, fisura centroalveolar, con mal pronóstico reconstructivo y neuromotor.
El Síndrome de Pai puede encontrarse también en combinación con alopecia frontal triangular, hipertelorismo discreto o hipotelorismo (2,8,10), nariz bífida, paladar alto, úvula bífida, diastema de la dentición maxilar, frenillo maxilar doble, clinodactilia, hernia inguinal, criptorquidia e hipospadias, entre otros. Lederle y col. (2) refieren en 1 de sus casos la presencia de un bronquio ectópico que surge de la tráquea a menos de 2 cm por encima de la carina.
La incidencia de este síndrome se estima en 1 de cada 20.000 a 40.000 recién nacidos, con mayor frecuencia en el sexo femenino (6). Su causa se desconoce (1-15) y su presentación es esporádica. Puede ser el resultado de anomalías cromosómicas y medioambientales. Masuno y col. (5) señalan como causa una traslocación de novo y describen a una familia con defectos de la línea media en 5 generaciones, comportándose con un patrón hereditario autosómico dominante de expresión variable, así como recesiva ligada al cromosoma X (4,5,9,15).
Caso clínico
Varón de 3 meses de edad producto de quinta gesta, madre de 34 años de edad y padre de 39, no cosanguíneos. Antecedente de tía materna con epilepsia y de hermano mayor de 6 años con autismo. Embarazo normoevolutivo y nacimiento a termino mediante cesárea por presentación pélvica, sin complicaciones postnatales.
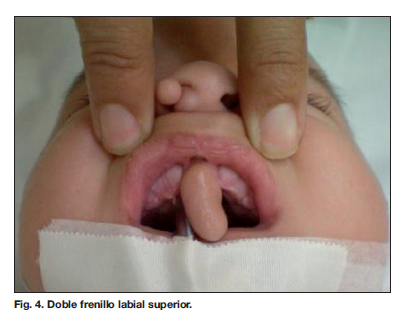
Es referido a la consulta externa de Cirugía Plástica Pediátrica por presentar dificultad respiratoria nasal por obstrucción de la narina derecha por pólipo, así como masa intraoral pediculada en la encía superior entre 2 frenillos labiales (Fig.1-4). Normocéfalo, sin hipertelorismo, epifora del ojo derecho, paladar alto, úvula y paladar bien formados.



La tomografía axial computarizada (TAC) de cráneo y macizo facial reveló agenesia de cuerpo calloso aunada a una imagen hipodensa en la línea interhemisférica a nivel de las astas frontales, correspondiente a un lipoma con bordes irregulares de 3,8 x 2,1 x 1,7 cm; espina bífida oculta a nivel de columna cervical y fisura central a nivel de la arcada del maxilar superior (Fig. 5-7).


El electroencefalograma estuvo dentro de los límites normales para la edad del paciene, sin evidencia de actividad paroxística ni de foco de lesión.
Practicamos resección quirúrgica en huso tanto del pólipo nasal único (de forma irregular, lobulado que midió 0,9 x 0,7x 0,5 cm) como del pólipo intraoral (de 2 x 1 x 0,7 cm) (Fig.8-10). El estudio anatomopatológico informó de pólipos escamosos queratinizados con presencia de anexos pilosebáceos y de tejido fibroadiposo, con escasas glándulas mucinosas sin componentes inmaduros o malignos.


El seguimiento del paciente en consulta externa hasta la edad de 1 año y 8 meses en el momento de la publicación de este artículo, ha sido favorable y sin complicaciones. Neurológicamente, a pesar de presentar agenesia del cuerpo calloso, se mantiene alerta con desarrollo sicomotor acorde para su edad y sin crisis convulsivas (Fig. 11 y 12). El estudio cromosómico fue masculino 46 XY con cariotipo normal.


Discusión
A los 27 años de su descripción inicial, Pai (1) presentó este síndrome en 1987 como una variante de la displasia fronto-nasal y desde entonces, solo se han documentado de forma esporádica 38 casos (7). Sus semejanzas y diferencias se deben a la variabilidad fenotípica (2-5) y se resumen en las Tablas I y II.


El caso que presentamos es de un varón. El estudio cromosómico fue 46XY sin cambios estructurales y posiblemente como un caso aislado sin antecedentes heredofamiliares. La prevalencia por género es mayor en el sexo femenino según Vacarrella (6). De los 38 casos publicados, 22 son femeninos (58%) y 16 masculinos (42%).
En nuestro paciente no se llegó al diagnostico de Síndrome de Pai hasta el nacimiento, y no por ultrasonido prenatal como otros casos publicados (13%) por Chousta (11), Andreeva (12) Oack (13) y Boulet (14).
Los pólipos frontonasales, en diferentes combinaciones, (nasales 25/38=66%, cutáneos 27/38=71% y 12/38=31% alveolares) se encontraron en el 100% de los casos documentados. En el caso que presentamos se dio la combinación de 1 pólipo nasal congénito y 1 maxilar entre 2 frenillos labiales, sin presentación de pólipos cutáneos ni de fisura del labio superior (52%). El pólipo nasal le ocasionaba dificultad para respirar por la nariz por obstrucción y el pólipo alveolar dificultad para la alimentación; una vez resecados quirúrgicamente cedieron estos síntomas, evolucionando favorablemente. La descripción patológica para ambos pólipos fue de tumoraciones dermoides o hamartomas, coincidente con lo referido en la literatura al respecto (3,6,8).
La presencia de un lipoma intracraneal en esta zona se presentó en el 84% de los casos descritos, con afección del cuerpo calloso en el 57% (Tablas I y II). En nuestro caso se diagnosticó agenesia del cuerpo calloso con lipoma intracraneal. El estudio de electroencefalografía fue normal y sin repercusión neuromotora hasta el momento actual.
Dentro de la gama de posibles combinaciones en la presentación de este síndrome, nuestro caso es muy semejante al recogido por Zenetta y col. (3). Ambos de sexo masculino, caracterizados por un pólipo nasal en la narina derecha y otro en región alveolar entre 2 frenillos labiales, sin pólipos cutáneos y sin labio hendido, presencia de lipoma intracraneal y agenesia del cuerpo calloso.
Conclusiones
A los 27 años de su primera descripción, el Síndrome de Pai cuenta con tan solo 38 casos recogidos en la literatura mundial. En la actualidad, con las técnicas de ultrasonido en 3D durante el embarazo, es posible sospechar el cuadro clínico que se comprueba al nacimiento.
Consideramos nuestro caso como el primero recogido en México que cumple con los criterios para ser diagnosticado como Síndrome de Pai.
Ante toda masa tumoral pediculada en la línea media facial se recomienda descartar extensión intracraneal con la finalidad de advertir a los padres de un posible retraso psicomotor o inclusive, el origen de crisis convulsivas.
Bibliografía
1. Pai JS, Levkoff AH, Leithiser RE Jr: Median cleft of the upper lip associated with lipomas of the central nervous system and cutaneous polyps. Am J Med Genet 1087, 26:921-924. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dra. Laura Melloni Magnelli
Hospital Ángeles Valle Oriente
Frida Kahlo 180-317, San Pedro
Valle Oricate, San Pedro Garza García
CP 66260 Monterrey, Nuevo León, México
e-mail: lmelloni@hotmail.com

