










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El síndrome de Moebius (SM) es una enfermedad congénita caracterizada por parálisis facial congénita no progresiva, simétrica o asimétrica, con ausencia de abducción de los ojos debida a lesión del VI y VII nervios craneales, en asociación con otras alteraciones neurológicas, músculo-esqueléticas, craneofaciales, respiratorias, y ortopédicas, entre otras. Se trata de un trastorno congénito raro, por lo cual no es fácil encontrar datos estadísticos respecto a su incidencia, que se calcula en torno a 1 caso por cada 50.000 nacidos vivos.
La parálisis congénita del nervio facial y abducens fueron descritas por Von Graefe en 1880, seguido por Harlam en 1881 y Christholm en 1882. Sin embargo, fue un neurólogo alemán, Paul Julius Moebius, quien notó la asociación de dicha parálisis facial congénita con otras malformaciones y realizó una comparación con casos previamente descritos, elaborando la clásica descripción del síndrome inicialmente conocido como Infantile Kernschwund y que posteriormente llevó su nombre.
En los últimos años se han asociado una gran variedad de malofrmaciones con este síndrome, lo que ha hecho de él un síndrome con alta variabilidad, con diferentes presentaciones, sin una etiología o patología bien definida. Existen 3 etiologías propuestas por 3 autores diferentes para tratar de entender la enfermedad:
•Legum y col. propusieron un defecto genético en el desarrollo del romboencéfalo, incluyendo el origen del VII nervio craneal.(1)
•Bavinck y Weaver describieron una interrupción del aporte vascular en el territorio de la arteria subclavia entre la sexta y séptima semanas de gestación, con isquemia del núcleo de nervio facial.(2)
•Por último, la exposición a teratógenos durante el embarazo puede ser causa de las 2 hipótesis previas. El ejemplo más claro es el uso como abortivo no-electivo del misoprostol, un análogo prostaglandínico E1, durante el primer trimestre del embarazo.
Debido a la heterogenicidad del síndrome se han propuesto diferentes clasificaciones para unificar criterios. Una muy conocida es la publicada por Abramson, Cohen y Mulliken quienes utilizan el acrónimo CLUFT, por sus siglas en inglés, para malformaciones en el nervio craneal (craneal nerve), extremidad inferior (lower limb), extremidad superior (upper limb), cara (face) y tórax (thorax).(3) Dentro de las malformaciones craneofaciales se han descrito: telecanto, microgenia o micrognatia, defectos del pabellón auricular y paladar hendido, entre otras. Las malformaciones de tipo músculo-esqueléticas abarcan: sindactilia, mano hendida, pie equino varo y síndrome de Poland como las más frecuentes. De la misma forma, hay malformaciones oftalmológicas com estrabismo y defectos de refracción, y neurológicas como epilepsia y calcificaciones cerebrales.
En el año 2002, la Dra. Terzis propuso una nueva clasificación que permite al cirujano identificar deficiencias en los pacientes y establecer una propuesta quirúrgica:(4)
–Grupo A: conocido como Síndrome de Moebius Clásico, con parálisis facial bilateral completa y parálisis del nervio abducens.
–Grupo B: o Síndrome de Moebius Incompleto, caracterizado por parálisis facial congénita con movimiento residual unilateral.
–Grupo C: conocido como Síndrome de Moebius-Like o similar a Moebius, en donde los pacientes tienen parálisis facial unilateral pero otros nervios craneales afectados.
En el departamento de genética, el SM frecuentemente se ha reportado como una patología esporádica; sin embargo, algunos casos se han asociado con herencia autosómica dominante, recesiva o ligada al X. Estudios citogenético de bandas GTG sugieren alteraciones en dos loci: 13q12.2-13 y 1p22.(5,6)
En México, según nuestra revisión, existen solo 5 publicaciones recientes de Síndrome de Moebius. Tres de ellas únicamente presentadas como reporte de casos en los estados de México, Tamaulipas y Tabasco, sin tener estadísticas concretas de la patología a nivel nacional.(7,8,9) Otra por la Dra. Borbolla y col. con una serie de casos en un centro hospitalario de atención pediátrica enfocado al estudio oftalmológico.(10) Y finalmente, otro publicado este año por nuestro equipo con respecto al manejo quirúrgico.(11)
El estudio que ahora presentamos es el resultado de la formación de un equipo multidisciplinario en nuestro centro hospitalario con la intención de estudiar la enfermedad para entenderla mejor, describir el espectro tan grande de la misma, y ofrecer los tratamientos más oportunos a cada uno de nuestros pacientes.
Material y método
Realizamos un estudio descriptivo, observacional, ambispectivo de todos los pacientes, tanto pediátricos como adultos, atendidos en el Hospital General “Dr. Manuel Gea González” de la Ciudad de México (México) con diagnóstico de síndrome de Moebius desde el año 2013 hasta 2017, en sus 3 presentaciones, confirmados clínicamente y ratificados por electromiografía, con la intención de lograr describir la gran variabilidad de presentación clínica del síndrome así como identificar alteraciones de imagen, cariotipo o estudio molecular de los pacientes.
Todos los pacientes fueron sometidos a exploración física detallada por parte del equipo multidisciplinario compuesto por cirujanos plásticos, oftalmólogos, otorrino laringólogos, foniatras, neurólogos, pediatras, ortopedistas y genetistas, y en conjunto analizamos estudios de imagen así como expedientes clínicos. De la misma forma, a todos los pacientes se les realizó cariotipo y estudio molecular por parte del equipo de genética con apoyo del Instituto Nacional de Medicina Genómica (INMeGen).
De acuerdo a las consideraciones éticas de nuestro país, todos los procedimientos estuvieron de acuerdo con lo estipulado en el Reglamento de la ley General de Salud en Materia de Investigación para la Salud, título segundo, capítulo I, Artículo 17, Sección II, investigación con riesgo mayor al mínimo para toma de muestras.(12)
RESULTADOS
Incluimos en el estudio 115 pacientes que contaban ya con el diagnóstico de síndrome de Moebius o sus variantes. De estos, 63 pacientes (54.7%) fueron mujeres y 52 (45.3%) varones, con edades que vairaban desde los 6 meses a los 40 años (media: 11.7 años).
De acuerdo a las definiciones establecidas previamente y en base a la clasificación propuesta por la Dra. Terzis, 69 pacientes (60%) fueron diagnósticados como síndrome de Moebius Clásico o Completo, 39 pacientes (34%) como síndrome de Moebius Incompleto, y solo 7 pacientes (6%) como síndrome de Mobius-Like. Presentamos las características demográficas del grupo de estudio en la Tabla I y en la Fig.1.
Tabla I Características demográficas de los pacientes de nuestro grupo de estudio
| Síndromede Moebius | Síndrome Moebius Incompleto | Síndrome de Moebius-Like | |
|---|---|---|---|
| N° de pacientes | 69/115 (60%) | 39/115 (34%) | 7/115 (6%) |
| Género Mujeres |
50 (43.4%) | 15 (13%) | 7 (6%) |
| Hombres | 19 (16.5%) | 24 (20.8%) | 0 (0%) |
| Cariotipo | 68 normales 1 translocacióncromosoma 4 a 10 | 39 normales | 7 normales |
| Consumo maternode misoprostol | 11/115 (9.5%) | 4/115 (3.4%) | 1/115 (0.8%) |

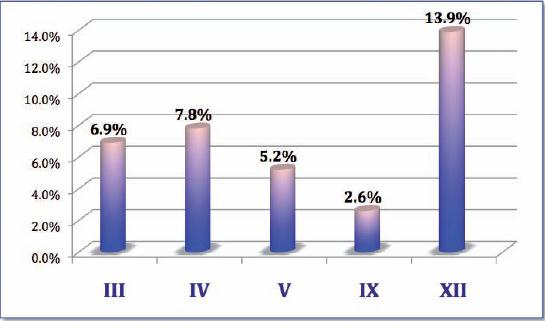
Al evaluar los estudio electromiográficos previos de los pacientes encontramos que los nervios más afectados además de VII y VI nervios craneales (NC) fueron el hipogloso o XII NC en el 13.9% (n= 16) de los pacientes, seguido por el IV NC en el 7.8% (n= 9), III en el 6.9%(n= 8), V en el 5.2% (n= 6) y el IX en el 2.6% (n= 3) (Fig. 2).
Recogimos las manifestaciones clínicas más frecuentes así como los hallazgos aislados, y los clasificamos en diferentes categorias: (Fig. 3 y 4)
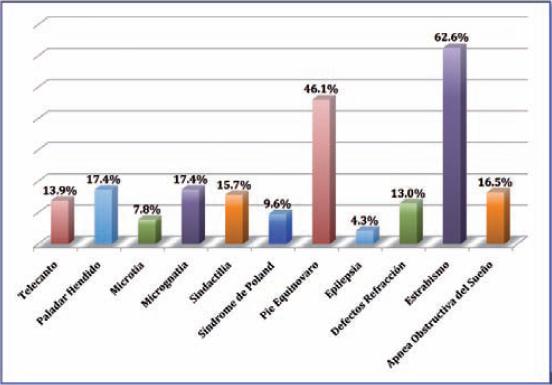
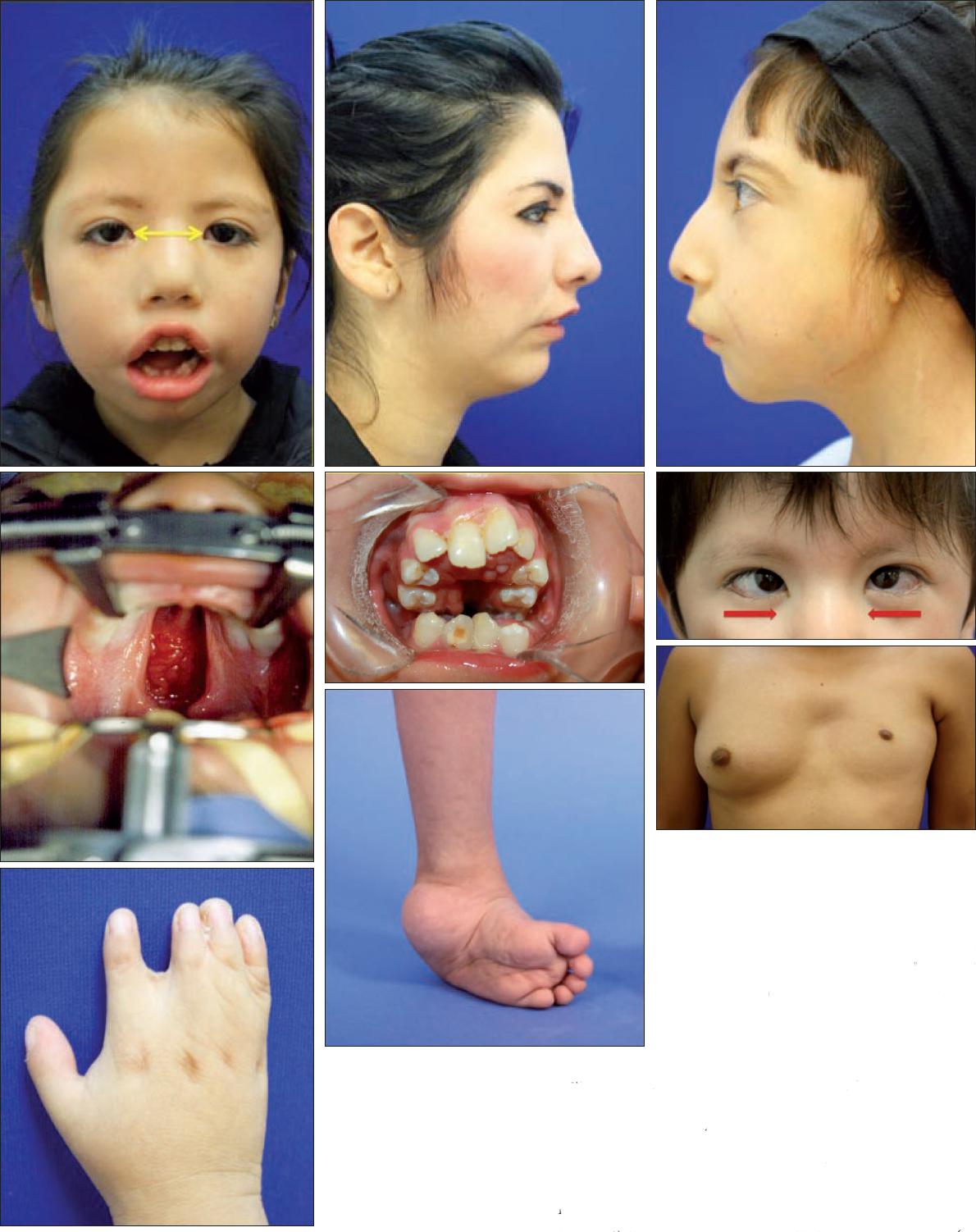
Fig. 4 Manifestaciones frecuentes asociadas al síndrome de Moebius: a) telecanto y pliegues epicantales; b) micrognatia y microgenia; c) microtia; d) alteraciones oclusales; e) paladar hendido y amelogénesis imperfecta; f) estrabismo; g) sindactilia; h) pie equino varo; i) síndrome de Poland.
–Craneofaciales. De las manifestaciones craneofaciales las más frecuentemente observadas fue el telecanto en 16 pacientes (13.9%), pero encontramos que 20 pacientes (17.4%) presentaban paladar hendido asociado, otros 20 micrognatia (17.4%) y 9 (7.8%) alteraciones del pabellón auricular.
–Musculoesqueléticas. Un total de 53 pacientes (46.1%) presentaron pie equino varo, siendo esta la patología de extremidades más frecuente; 18 (15.7%) tenian algún grado de sindactilia; y 11 (9.6%) síndrome de Poland asociado. En este grupo de pacientes solo se reportó 1 caso de escoliosis tóraco-lumbar, 1 caso de agenesia de pie derecho y 1 caso de mano hendida atípica bilateral.
–Neurológicas. En este apartado, además de mencionar los nervios craneales comprometidos, encontramos 5 casos (4.3%) de epilepsia. Un equipo psiquiátrico realizó exámen mental a estos pacientes, reportando 3 casos con retraso mental.
–Oftálmológicas. De las manifestaciones oftálmicas, 72 pacientes (62.6%) presentaron estrabismo convergente y 15 (13%) tenían algún defecto refractivo como hipermetropia, astigmatismo o miopía, aunque en pacientes muy pequeños este dato es difícil de valorar.
–Ortodóncicas. Aparte de los casos mencionados de micrognatia, documentamos 1 solo caso de anquilosis de la articulación témporo-mandibular asociada.
–Genéticas. En este apartado realizamos en total 115 cariotipos, de los cuales encontramos 114 (99.1%) normales y 1 solo caso (0.9%) con translocación recíproca balanceada entre el cromosoma 4 y el cromosoma 10 que involucra los puntos de ruptura 4q21 y 10q11.2 (Fig. 5 ). La misma transolocación se encontró en la madre de la paciente. También 16 casos (13.9%) en los que la madre confirmó haber ingerido misoprostol en el primer trimestre del embarazo. Enviamos las muestras al INMeGen para análisis molecular, pero por falta de recursos materiales no se pudieron procesar.

Fig. 5 A y B. Cariotipo de paciente con síndrome de Moebius y translocación recíproca balanceada entre cromosoma 4 y 10. Madre e hija con la misma alteración.
–Imagen. Identificamos 1 caso de sobrecrecimiento del cuarto ventrículo cerebral con ausencia parcial de vermix cerebelar, conocido como síndrome de Dandy-Walker. De forma aislada recogimos 1 caso de braquicefalia, 1 caso de hipoplasia de tercio medio facial y 1 caso de plagiocefalia anterior; estos últimos 3 casos confirmados inicialmente por la exploración física.
–Otras. Encontramos, de forma aislada, 1 solo caso de amelogénesis imperfecta asociada al SM y 1 caso de esferocitosis asociada. En apoyo con el Servicio de Otorrinolaringología llevamos a cabo polisomnografías a los pacientes, encontrando 19 casos de apnea obstructiva del sueño.
Discusión
El síndrome de Moebius es una patología congénita rara, poco entendida y que requiere un abordaje multidisciplinario para su estudio y tratamiento ya que puede estar asociado a múltiples alteraciones. Por esta razón, en nuestro centro hospitalario, logramos establecer un equipo multidisciplinario formado por cirujanos plásticos, oftalmólogos, otorrinolaringólogos, foniatras, neurólogos, pediatras, ortopedistas y genetistas, para abordar y tratar a nuestros pacientes de forma integral.
La serie más actual y más extensa en el siglo XXI era la serie italiana publicada por Arturo Carta en el 2011, donde reúne 55 pacientes para realizar una descripción de las manifestaciones oftálmicas, principalmente.(13) En 2014, Borbolla y col. del Servicio de Oftalmología Pediátrica del Instituto Nacional de Pediatría de nuestro país publicaron una serie de 64 casos en la que describen principalmente manifestaciones oftalmológicas, siendo la serie más grande publicada hoy en día.(10) En nuestra serie, junto con el apoyo de múltiples servicios dentro de un solo centro hospitalario, logramos identificar 115 casos así como las manifestaciones clínicas presentadas en cada uno de ellos.
El equipo de John Mulliken y col., en 1998(3) publican una serie de 27 pacientes en una sola unidad hospitalaria y proponen la nemotecnia CLUFT por las siglas en inglés cranial nerve, lower limb, upper limb, face and thorax malformation para el estudio de estos pacientes, con la intención de lograr una integración para su estudio de forma interinstitucional y homogeneizar los términos. De forma similar a lo reportado por nosotros, sus pacientes tenían compromiso del VII y del VI nervios craneales. Tanto el estudio de Mulliken como el de Terzis en el 2002(4) coinciden en que junto con la parálisis facial, los pares craneales V y XII son los más afectados; seguimos por los demás en procentajes similares. Estas cifras contrastan con nuestros resultados en donde el nervio hipogloso (XII) y el patético/troclear (IV) son los más afectados en el 13.9% y el 7.8% respectivamente, pero con diferencia en el número de casos entre los 27 de Mulliken, los 43 de Terzis y los 115 de nuestra serie.
Tal y como describen la mayoría de las publicaciones en la literatura, como las de Terzis, Carta y Ramos de Souza-Dias,(14) el estrabismo fue la manifestación clínica más frecuente en el 62.6% de los casos y los defectos refractivos aparecieron en el 13% de los casos, aunque muchos de los pacientes no pudieron ser valorados por su corta edad. También coincidimos en que las manifestaciones craneofaciales más frecuentes son el telecanto, el paladar hendido y el pie equino varo como manifestación musculoesquelética más frecuente en el 46.1% de los casos, seguido de la sindactilia y del síndrome de Poland, entre otros. El síndrome de Moebius asociado a craneosinostosis no sindrómica, esferocitosis y amelogénesis imperfecta, no había sido reportado previamente en la literatura.
La relación del consumo de misoprostol con el diagnóstico de síndrome de Moebius sigue siendo un tema de incertidumbre e investigación. En 1998, Pastuszak y col.(15) en un estudio multicéntrico de 96 pacientes describen que 47 (49%) tuvieron madres que tomaron misoprostol durante el primer trimestre del embarazo. Aunque no fue motivo de nuestro estudio, encontramos que 16 pacientes (13.9%) tuvieron madres que tomaron misoprostol en el primer trimestre, los que aunque no es similar al trabajo brasileño, hemos de tener en cuenta que en muchos casos las madres no confiesan el consumo en las primeras consultas. En estos 16 pacientes existe una diferencia importante en la asociación con los 3 tipos de presentación del síndrome, siendo más frecuente la asociación con el síndrome de Moebius Clásico en el 68.7% y tan solo en el 25% con síndrome de Moebius Incompleto y en el 6.3% con síndrome de Moebius-Like.
Por otra parte, la mayoría de las publicaciones de síndrome de Moebius hacen referencia a que no hay evidencia suficiente para apoyar un patrón de herencia autosómico dominante, sino más bien mutaciones de novo. Ziter y col.(5) en 1977 presentaron una translocación recíproca entre el cromosoma 1p34 y el cromosoma 13q13, así como Slee y col(6) en 1991 quienes publican una deleción en el cromosoma 13q22.2, lo que suponía que el daño podría estar en el cromosoma 13q22.2 - q13, por lo que Uzumcu y col. en 2009, realizan un estudio molecular para buscar esas mutaciones en 9 pacientes sin encontrarlas. En nuestro estudio encontramos una translocación no antes reportada entre el cromosoma 4 y el 10, desafortunadamente sin tener resultados del estudio molecular.
La serie de pacientes que presentamos tiene la gran ventaja de pertenecer a un solo hospital con la oportunidad de poder ofrecer al paciente tratamiento por el mismo grupo multidisciplinario de especialistas, hecho fundamental que permitirá diversas líneas de investigación con otras instituciones, asi como profundizar más sobre la fisiopatología de este síndrome con la posibilidad de identificar asociaciones causales específicas del mismo. Por otro lado, con el apoyo del Instituto Nacional de Neurología mexicano, planeamos realizar estudios de resonancia magnética cerebral funcional en combinación con tomografías con emisión de positrones con la realización de diferentes paradigmas, como movimientos de lengua, de labio, sonrisa, movimientos de mejilla, parpadeo ocular y deglución, para identificar su integración en la corteza cerebral. En el área de la Cirugía Plástica, la cirugía para protección ocular y la cirugía para la sonrisa son los protocolos de tratamiento actual, y estamos realizando un análisis de cada uno de estos procedimientos.
Conclusiones
Hasta la fecha, este trabajo presenta hasta donde hemos podido comprobar, la cohorte con pacientes vivos con síndrome de Moebius más extensa en un solo centro en nuestro país y en el mundo. Hemos logrado reunir 115 casos y pretendemos llegar a 150 casos en 1 año más.
Describimos las patologías asociadas a la enfermedad para que puedan ser abordadas por un equipo multidisciplinario y lograr así un tratamiento óptimo para cada uno de nuestros pacientes.

