










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkOncología (Barcelona)
versión impresa ISSN 0378-4835
Oncología (Barc.) vol.27 no.10 2004
ARTÍCULO ORIGINAL
Apoptosis y antiapoptosis en cáncer, Alzheimer y procesos neurodegenerativos: ¿una dialéctica de contrarios? Nuevo abanico de posibilidades terapéuticas y peligros potenciales
S. Harguindey
Centro Médico La Salud Vitoria
Dirección para correspondencia
RESUMEN
Los intricados caminos de señalización intracelular en la desregulación de la apoptosis, espontánea o inducida, se han convertido en las principales dianas terapéuticas en enfermedades que van desde las neoplásicas a los procesos neurodegenerotivos. A la vista de los tendencias actuales en investigación se puede afirmar que el fracaso en inducir una apoptosis selectiva en las células y tumores cancerosas (proapoptosis), y en sentido contrario, lo imposibilidad de evitar lo apoptosis espontánea en enfermedades neurodegenerativas como el Alzheimer (antiapoptosis), representan una raíz y árbol común pora dos ramas, o áreas de la medicina, creciendo en sentidos opuestos.
El enfoque aquí presentado es un intento de estimular una visión integrada de ambas ramas y áreas de investigación, oncología y neurología, en orden a mejorar la comprensión de sus naturalezas íntimas, tratando así de incrementar el entendimiento de las anormalidades básicas y clínicas de los procesos bajo estudio. A esto se puede acceder a través de un escalonamiento diferenciador de las posibilidades terapéuticas que se están abriendo en la actualidad así como merced o una consideración preliminar de los peligros potenciales que pueden surgir en el empeño.
Palabras clave: Apoptosis. Antiapoptosis. pH. Cáncer. Enfermedad de Alzheimer. Enfermedades neurodegenerativas. p53. Factores de crecimiento.
SUMMARY
The intricated pathways of intracellular signaling and the deregulation of cell apoptosis, either spontaneous or induced, are becoming the main molecular and therapeutic targets ranging from cancer to neurodegenerative diseases. Modern trends in research and treatment show that the failure to induce selective cancer cell and tamor apoptosis (proapoptosis), and, in the opposite direction, the failure to prevent the spontaneous apoptosis of neurodegenerative diseases, like Alzheimer's disease, can be interpreted as problems representing two branches, or areas, of medicine growing in an opposite direction.
The approach here advanced represents an attempt to stimulate an integral vision of both branches or areas of research, oncology and neurology, hoping to improve the understanding of the intimate nature of the basic and clinical abnormalities under investigation. A stop by step differentiation of the therapeutic possibilities that are being opened nowadays is considered, as well as some preliminary and potential dangers raised by the very same treatment efforts.
Key words: Apoptosis. Antiapoptosis. pH. Cancer. Alzheimerts disease. Neurodegenerative diseases. p53. Growth factors.
Introducción
La apoptosis, muerte o suicidio celular programado, es un mecanismo esencial en la homeostasis celular, y así en la regulación fisiológica del organismo. La desregulación de la apoptosis por mecanismos, genética o medioambientalmente inducidos, conduce, por el contrario, a una variedad de patologías, ya sea como proapoptosis, o pérdida celular excesiva (enfermedades autoinmunes y neurodegenerativas), o por antiapoptosis, o acumulación celular exagerada (cáncer)1. La intricada y variada maquinaria celular de este proceso crea ciertos hechos característicos de la apoptosis, tales como encogimiento celular, condensación citoplasmática, formación de ampollas con hinchazón en la membrana, condensación de cromatina y fragmentación del DNA cromosómico, aunque el acontecimiento central a nivel molecular es la activación de ciertas proteasas cisteína/aspartato o caspasas (caspasas 3, 8 y 9 principalmente)2, 3.
La muerte celular en ciertas enfermedades degenerativas del sistema nervioso central (SNC) está relacionada, desde el punto de vista de la etiopatogenia, con la instauración de un proceso irreversible y espontáneo de apoptosis celular progresiva4-6. Por una parte, es generalmente aceptado que una apoptosis inapropiada contribuye a la pérdida neuronal en la mayoría de las enfermedades neurodegenerativas humanas (ENDs)5-7. En consecuencia, una opción terapéutica en procesos como la enfermedad de Alzheimer y otros procesos neurodegenerativos de etiología y fisiopatología confusas consistiría en interrumpir la cadena de señales celulares y/o la actividad de genes o productos génicos que conectan un daño neuronal, probablemente multifactorial, con los diferentes mecanismos proapoptóticos3. Por estas razones, ciertas caspasas y su inhibición son consideradas cada vez con mayor rigor como dianas terapéuticas de gran valor potencial en estos procesos degenerativos del sistema nervioso8.
Una situación diametralmente opuesta a ésta tiene lugar en el tratamiento de las enfermedades neoplásicas. Aquí, es precisamente el fracaso en la inducción de apoptosis celular, como muerte celular selectiva, lo que constituye una de las causas principales, si no la fundamental, de la resistencia a la quimioterapia de los tumores malignos y leucemias9,10. De ahí que muchos esfuerzos en estas líneas estén dirigidos a hallar estímulos para la inducción de una apoptosis celular selectiva como estrategia de búsqueda de dianas intracelulares6, 11, así como designar estrategias que permitan superar la resistencia a los inhibidores conocidos de proteino quinasas (PKs) utilizados en la actualidad10 (Tabla I).
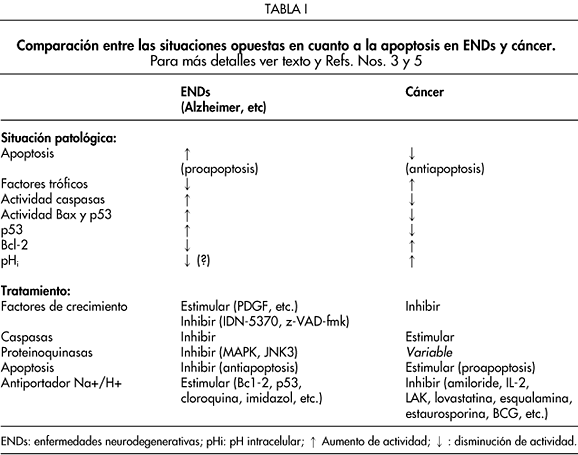
Buscando un sendero común para estos dos caminos opuestos, aunque entrelazados por sus efectos contrarios sobre una muerte celular inapropiada y espontánea, o la resistencia a ella, se hace comprensible considerar que ciertos factores de crecimiento sobreexpresados en algunas enfermedades neoplásicas podrían ser de utilidad en impedir o dificultar la apoptosis celular en la enfermedad de Alzheimer (EA), así como en otras enfermedades degenerativas del sistema nervioso central: (enfermedad de Huntington (EH), esclerosis lateral amiotrófica (ELA), esclerosis múltiple (EM), enfermedad de Parkinson (EP) y retinitis pigmentaria (RP)3,12. Al mismo tiempo, estas nuevas vías y aproximaciones conceptuales pueden hacer avanzar nuestro entendimiento y conocimiento de cómo inhibir más efectiva y selectivamente la acción de oncogenes y factores de crecimiento en un intento de matar selectivamente por inanición metabólica a nivel molecular, a las células y tumores malignos.
I) Un lado de la moneda: inhibición de apoptosis como tratamiento de la enfermedad de Alzheimer y otros procesos neurodegenerativos
La apoptosis celular progresiva es causa fundamental de pérdida neuronal en la mayoría de las enfermedades neurodegenerativas humanas4-8. Dicho tipo de muerte celular, diferente de la necrosis, puede inducirse bien por 1) la estimulación activa de mecanismos que induzcan este tipo de muerte celular (caspasas)6, 8, o bien, 2) por medio de la depleción espontánea o retirada activa de ciertos factores de crecimiento tróficos en diversos sistemas celulares (thropic factor withdrawal)13 (Tabla I). En consecuencia, pero en la dirección opuesta, las dos aproximaciones antiapoptóticas y terapéuticas a estos procesos neurodegenerativos pueden ser, respectivamente, A) Inhibiendo los mecanismos que inducen pérdida neuronal por estimulación anormal de las dianas de señalización intracelular de ciertas caspasas, y B) Tratando de impedir una apoptosis patológicamente aumentada por medio de una falta de estimulación trófica con la utilización de factores de crecimiento. Ambas posibilidades están siendo consideradas ampliamente en la actualidad por diversos grupos y en una multiplicidad de ensayos clínicos y preclínicos14.
En cuanto a la primera vía terapéutica, (vía A), las caspasas son proteasas intracelulares que participan en los caminos apoptóticos de las células humanas, incluidas las neuronas. La inhibición de dichas caspasas (caspasas 3, 4 y 9, principalmente) se ha mostrado útil en evitar la muerte celular neuronal programada en modelos in vitro e in vivo de enfermedad neurológica. Dichas caspasas parecen estar involucradas en pasos tempranos de disfunción neuronal y agregación de proteínas en enfermedades como el Alzheimer y la enfermedad de Huntington, estando también involucradas en la patogenia de la enfermedad de ParLinson y en la esclerosis lateral amiotrófica (ELA)3.
En cuanto a la enfermedad de Alzheimer, se sabe que la acumulación de B-amiloide es causa de muerte celular por apoptosis inducida por activación de las caspasas. Por ello, ciertos inhibidores formacológicos de caspasas, como BAF, z-VAD-FMK, etc, son capaces de inhibir la muerte neuronal. Lamentablemente, estudios in vivo en esta enfermedad son prácticamente imposibles de ser llevados a cabo, por razones obvias, y en los limitados modelos existentes la inhibición de caspasas sólo es capaz de logar una inhibición débil y transitoria de la apoptosis y otros fenómenos neurodegenerativos, y aún así, no en todos los modelos estudiados3-5.
De todos modos, la inhibición selectiva de caspasas podría reducir la apoptosis neuronal en enfermedades neurodegenerativas. Una sobreexpresión, o niveles aumentados de moléculas proapoptóticas como Bax y p53, junto con una inhibición de la anti-apoptótica proteína Bc1-2, aparte de otros factores, han sido implicados en la apoptosis de la enfermedad de Alzheimer, en Parkinson y en esclerosis lateral amiotrófica (Tabla I). La quinasa terminal jun-N ha sido asimismo involucrada en estos procesos neurodegenerativos4, 12. Paralelamente, células neuronales que sobreexpresan la proteína anti-apoptótica Bc1-2 están protegidas contra la apoptosis inducida por taxol o ácido okadaico, un efecto mediado por sus efectos sobre el citoesqueleto neuronal15, 16. Como se verá más adelante, estas anomalías son diametralmente opuestas a las que tienen lugar en la patológica resistencia a la apoptosis típica de las enfermedades neoplásicas, ya sean tumores malignos o leucemias (Tabla I).
En la misma línea de terapéutica antiapoptótica, la inhibición, genética o farmacológicamente inducida del gen supresor p53, por otra parte factor clave en favorecer tanto la transformación neoplásica17 como el crecimiento tumoral, y a su vez determinante en la resistencia a la terapéutica antineoplásica18, se muestra como neuroprotectora. Estos hechos hacen lógico el pensamiento de Waldeimer cuando afirma que, debido a que una funcionalidad normal de p53 presenta actividad anticancerosa y supresora de la oncogénesis, su inhibición en enfermedades como PD o AD podría ser problemática por la posibilidad de inducción carcinogenética y posterior estimulación del crecimiento tumoral4. En este sentido, es muy importante señalar que el mecanismo molecular recientemente implicado en la pérdida de la función protectora del p53 y subsiguiente carcinogénesis está relacionado con un aumento el pH intracelular, mientras que, por el contrario, un microombiente acídico intracelular parece una condición sine qua non para facilitar la apoptosis inducida por el p5319. Este último hallazgo indica un hecho fundamental: que el equilibrio ácido-básico de la célula, tumoral o degenerativa, es el mecanismo molecular esencial que determina la actividad, normal o alterada, del gen supresor p53, como ya ha sido sugerido con antelación por Di Giammarino y cols, entre otros17, 19.
De forma paralela, en la actualidad se están estudiando una serie de inhibidores de la JNK, un miembro de la familia de quinasas MAPK, debido a la evidencia existente de un potencial terapéutico para inkibidores de MAPK, como el CEP-11347, en antagonizar la formación de B-amiloide (AB), fenómeno a su vez inducido por la activación de JNK12, 13. Esta activación y/o sobrexpresión secundaria a la activación de la JNK3, que también puede ser inducida por Ab, parece tener un papel clave en disparar la cascada apoptótica en enfermedades neurodegenerativas. Por todo ello, se considera que inhibidores selectivos de JNK3 podrían presentar un efecto terapéutico en el tratamiento de Alzhelmer y/o la esclerosis múltiple, etc., pero que, desafortunadamente, también podrían estimular la hiperproliferación celular y la transformación neoplásica12.
La segunda vía terapéutica antes considerada (vía B), o apoptosis por deprivación de factores tróficos y/o de crecimiento, como el factor de crecimiento neuronal o NGF, u otros, representa asimismo un mecanismo alternativo para la activación, por un defecto que se torna como estimulador de la apoptosis, de la vía de las caspasas, lo que a su vez precede la muerte celular neuronal14 (Tabla I). Se ha sugerido que una inadecuada producción de moléculas tróficas por los mismos tejidos nerviosos, o defectos de ciertos factores de crecimiento en el microambiente celular inducidos por causa sistémica, pueden constituir un factor significativo en la muerte celular14-15. Así se puede entender que una terapéutica antiapoptótica para varias de estas enfermedades degenerativas del sistema nervioso esté cada vez con más frecuencia basada en la aplicación de compuestos que estimulen el trofismo celular, ya sean propargilaminas14, activadores del receptor alpha-2-adrenérgico20, etc.
Hay que tener presente un hecho adicional de potencial importancia terapéutica, como el que entre las dianas celulares clásicas del PDGF se hallen las neuronas, las células gliales y las células epiteliales pigmentarias de la retina, por contener receptores a PDGF. Estas propiedades, aunque abren nuevas posibilidades de tratamiento en ciertas ENDs, al mismo tiempo advierten del peligro que existe de que dichas apuestas terapéuticas presenten ciertas contraindicaciones si se tiene en cuenta que los glioblastomas, como otros diversos tumores, presentan expresión de PDGFR-a y que la estimulación autocrina por PDGF contribuye tanto a la trasformación inicial como a la progresión tumoral de esta patología cerebral neoplásica21. La depleción o inhabilitación de factores de crecimiento como el IL-2, IL-3, etc, parece ser asimismo suficiente para inducir una apoptosis celular, considerada hasta ahora como "espontánea", debido exclusivamente al desconocimiento existente sobre su etiopatogenia, sobre todo en enfermedades neurodegenerativas tipo Alzheimer22.
Esto es asimismo fundamental desde el punto de vista terapéutico, ya que hoy en día existe la tecnología necesaria para la utilización clínica de factores de crecimiento autólogos en diversos procesos y especialidades médicas, ampliamente utilizados como inductores y estimuladores de la reparación acelerada de diversos tejidos23. En este sentido, es importante considerar al respecto que las principales vías pro y antiapoptóticas de señalización intracelular sean mayormente conocidas, existiendo incluso mapas explicativos de dichos caminos y senderos intracelulares así como sus posibles dianas terapéuticas6.
En cuanto a los mecanismos mediadores de la mayoría, si no de todos los factores de crecimiento en estos procesos, se encuentra una alteración en el intercambio celular de Na+ y H+ como mecanismo clave de intermediación, sugiriendo que, en su versión proapotótica aplicada a las ENDs, la señal para la acidificación intracelular es la misma que para la apoptosis22. La activación de caspasa-8, por ejemplo, es precedida por una acidificación intracelular en células inducidas a la apoptosis por somatostatina2, mientras que el inhibidor universal de las caspasas, z-VAD-fmk, previene la disminución del pHi, y así la apoptosis2, 3. Por el contrario, la acidificación intracelular per se activa las caspasas sensibles a la z-VAD-fmk2 y es precursora de la apoptosis24. En la misma línea patogenética, la sobreregulación o dimerización de proteínas BAX, así como la activación de caspasas son disparadas por un pHi acídico22. Para una revisión más amplia de las vías y mapas de señalización intracelular en la apoptosis mediada por las caspasas, ver refs. 1 y 20. Para un estudio más en profundidad sobre los aspectos moleculares de la inhibición de caspasas y antiapoptosis como estrategia terapéutica en enfermedades neurodegenerativas, ver refs. 3 y 5.
Finalmente, algunos inhibidores de ciertas caspasas involucradas en la maquinaria de activación de la apoptosis celular, como el IDN-5370, ejercen un efecto protector de la apoptosis en neuronas sinápticas y corticales. De manera paralela, el inhibidor universal de las caspasas, z-VAD-fmk, o la sobreexpresión de BCL-2 evitan la apoptosis22. No sólo el segundo, sino ambos mecanismos, aparecen como directamente relacionados con la acidificación intracelular que precede a la apoptosis en diferentes tipos de células, un proceso que a su vez se pone en marcha por Fas o receptores de somatostatina2, 16, 30, 31.
Asimismo, diferentes factores tróficos presentan una actividad antiapóptica, lo que está generalmente mediado por la estimulación del antiportador Na+/H+, con la consiguiente prevención de una caída en el pH intracelular18, 32. Entre estos factores se hallan, la familia antiapoptotica BCL-2 y la actividad de un p53 no mutado y funcionalmente sano. Existe una aparente contradicción y paradoja, sin embargo, en el hecho de que se haya publicado, aunque en un contexto diferente, que al menos tres quinasas serina/treonina: la proteino-quinasa C, la calmodulina- quinasa 11 y la MAPK, pueden estimular el antiportador Na+/H+ y por consiguiente, inducir alcalinización intracelular22, lo que aconsejoría estimular estas PKs en las ENDs, al contrario de lo publicado por otros35. (Tabla I, columna 2, etiquetado como "variable").
El efecto protector contra la apoptosis de un pHi elevado es también ampliamente reconocido tanto en la inducción de resistencia a la quimioterapia33, 34 como en la mediada por receptores y factores de crecimiento32, 35. Evidencia adicional que apoya la estrecha relación causa-efecto entre alteraciones del pH celular y la activación o inhibición de la apoptosis celular es aportada por el hecho de que una elevación del pHi mediada por el imidazol o la cloroquina previene la apoptosis mediada por Fas, mientras que la inducción de una alcalinización intracelular juega un papel fundamental en la supresión de la apoptosis por PMA, Il-3, factor de estimulación plaquetario, etc. Independientemente de la quinasa efectora, la alcalinización resultante es el factor crítico para la supervivencia celular16, 22 (Tabla I).
II) El otro lado de la moneda: inducción de apoptosis en el tratamiento del cáncer
Si en el tratamiento de las enfermedades neurodegenerativas, incluidas bajo unas muy apropiadas iniciales de NENDsH, las dos principales líneas de investigación eran, A) la inhibición de los mecanismos que estimulan las dianas de señalización intracelular de estimulación de ciertas caspasas, y en segundo término, B) la inhibición de la apoptosis por estimulación trófica mediada por la utilización de factores de crecimiento, en el tratamiento del cáncer, por el contrario, lo que se trata de hacer es, exactamente lo diametralmente opuesto. Es decir, 1) estimular mecanismos que disparen selectivamente la apoptosis celular, estén o no relacionados con la activación de caspasas36, y en segundo lugar, 2) inhibir a estimulación trófica mediante la inhibición o retirada de factores de crecimiento, etc13 (Tabla I).
Así como la deprivación de factores tróficos u otros mecanismos paralelos de inhibición o desestimulación del metabolismo y viabilidad celulares, favorecen la instauración de una suicida cascada apoptótica en ENDs, la dificultad para inducir una apoptosis selectiva en células y tumores malignos se tiene como una de la principales causas, si no la principal, del fracaso terapéutico en el tratamiento de muchas enfermedades neoplásicas en la actualidad11, 18, 22, 32. Por ello, una de las principales vías a las que apuntan terapéuticamente las más modernas vías de investigación terapéutica en oncología traslacional es la actuación inhibidora sobre proteinoquinasas activada por mitógenos (MAPK) por medio de inhibidores de dichas PKs (MEK), como imantib o gefitinib37, y posteriormente sobre otras dianas y mecanismos asociados que lleven a superar la resistencia a los inhibidores de las proteinoquinasas10, 38.
Por ello, se torna de fundamental importancia el llegar a conacer las vías y fenómenos universales mediadores en la apoptosis celular, ya sea en orden a inducirla (cáncer) o inhibirla (ENDs). Este enfoque puede posteriormente ayudar a reconocer las dianas y mecanismos de posible explotación terapéutica en unas y otras situcciones. Así, es un hecho reconocido que diversos productos capaces de inducir apoptosis en células cancerosas de diversas estirpes, tales como 11-2, LAK, lovastatina, estaurosporina y BCG ejercen su efecto terapéutico y apoptótico a través de la inhibición del mecanismo de membrana representado por el antiportador Na+/H+ y, consecuentemente a ello, por una acidificación intracelular16, 25, 28. El alcanzar una cierta disminución del pHi parece ser un factor esencial en permitir la iniciación de una reacción en cadena que acaba en la apoptosis28, 31. Por el contrario, cualquiera de estos compuestos tendría muchas probabilidades de presentar un efecto dañino y agravador en las enfermedades neurodegenerativas, o ENDs, que de por sí tienden a una pérdida celular progresiva por apoptosis espontánea.
Por tanto, es comprensible que inhibidores específicos del antiportador Na+/H+ de la serie amiloride, u otros compuestos en la misma línea terapéutica (endostatina, angiostatina, esqualamina, 2-metoxiestradiol, anti-VEGF Ab, talidomida, etc.)39-42 hayan sido crecientemente considerados durante los últimos años como agentes anticancerosos, mientras algunos de ellos se han mostrado asimismo efectivos a la hora de aumentar la actividad y especificidad (sinergia) de diversos medicamentos antineoplásicos32, 43, 44. Se ha demostrado que la acidificación intracelular por debajo de un límite fisiológico está correlacionada con la apoptosis después de tratamiento con etopósido, camptotecina, cicloheximida, lovastatina, paclitaxel, etc22, 29, 44. Finalmente, una serie de compuestos proapoptóticos, con las caracteristicas de ser todos ellos acidificadotes celulares, están siendo estudiados en una multiplicidad de intentos de superación de la MDR (resistencia múltiple a fármacos antineoplásicos), compuestos tales como verapamil, ciclosporina A, tamoxifen, amiodarona, bafilomicina A, nigericina, etc16. Finalmente, estudios de actividad-tiempo han demostrado que la acidificación intracelular que tiene lugar en la apoptosis es posterior de la actividad de la roteasalCE/CED-3, porque la inhibición de la proteasa con zVAD.fluoromethylketona (FMK) completamente previene la disminución de pHi, la digestión del DNA y otros acontecimientos asociados a la apoptosis46.
Por el contrario, la sobreestimulación del antiportador Na+/H+ representa el principal, pero no el único, mecanismo esencial tanto en el mantenimiento como en la elevación del pH intracelular18, 22, 32, 44, lo que ese correlaciona con los conocidos efectos carcinogénicos directos de la elevación del pHi 47, 48. Por medio de la elevación del pHi, este antiportador juega un papel de pivote y esencial en la información de una multiplicidad de señales intracelulares, en la cascada mitogénica y en la actividad de diversas hormonas, factores tróficos y de crecimiento. Asimismo, su actividad excesiva y sobreexpresión alcalinizante es crucial en la activación funcional de diferentes oncogenes, como ras y v-mos, en la supresión de la actividad antineoplásica del oncogen supresor p5317, 19, y en actuar como mediador de la transformación y proliferación celular y en la síntesis de DNA17, 32, 47. Se concluye que la inhibición de la apoptosis en células cancerosas de diversas estirpes puede ser completamente suprimida o bien induciendo una alcalinización celular o previniendo una acidificación citosólica22, 29, 46, 49.
III) Aplicación local de factores de crecimiento y cáncer
La aplicación de diversos factores de crecimiento, por inoculación local, infusión o autotransfusión autólogos, es hoy en día algo perfectamente factible en la práctica médica23. Por tanto, este nuevo campo de investigación puede ofrecer importantes perspectivas y potenciales en el tratamiento de enfermedades en las que existe una depleción o deprivación de factores tróficos y de crecimiento, como el Alzheimer, esclerosis múltiple, etc.
Factores de crecimiento celular y metabolitos activadores del trofismo, crecimiento y proliferación celular como PDGF (factor de crecimiento derivado de las plaquetas), TGF-b (factor de crecimiento transformante b1), bFGF (factor de crecimiento fibroblástico), VEGF (factor de crecimiento vasoendotelial), PDEGF (factor de crecimiento endotelial derivado de las plaquetas), IGF-1 (factor de crecimiento tipo-insulina-1) etc, así como citoquinas como PF4 y CD40L, están siendo utilizados con creciente asiduidad en diversas especialidades tanto médicas como quirúrgicas, sobre todo en el área de reparación acelerada de tejidos23. Por medio de sencillos procedimientos ya descritos, se consigue un concentrado de plasma autólogo rico en factores de crecimiento, o PDGFs. Este preparado contiene una significativa concentración de factores de crecimiento mitogénicos tales como PDGF, TGF-beta, VEGF, bFGF, PDEGF, IGF-1, etc. Al mismo tiempo, algunos de estos factores son conocidos por presentar un potente efecto estimulador, no sólo de la proliferación celular sino también de la angiogénesis, mecanismo éste último por el cual influyen terapéuticamente en la formación y reparación de tejidos dañados, o incluso ausentes.
Sin embargo, ha surgido recientemente la preocupación de si la aplicación local de un concentrado de estos factores de crecimiento podría contribuir, como efecto secundario indeseado, a la transformación celular, y así a un aumento de la incidencia de tumores malignos y a un crecimiento tumoral y diseminación acelerados, al menos en las áreas circundantes a las tratadas. En este sentido, es bien sabido que algunos de estos factores de crecimiento se comportan como oncogenes en diversos sistemas celulares y están asociados a la transformación celular maligna, al menos in vitro18, 32, 47.
Asimismo, tanto el crecimiento local como la diseminación metastática son procesos altamente dependientes de la angiogénesis tumoral, o neoangiogénesis39, 50. Por otra parte, el hecho de que las plaquetas sean el origen de VEGF y bFGF, dos factores altamente angiogénicos, ha llevado a postular que las plaquetas, por su capacidad de liberar factores relacionados con la neoangiogénesis, podrían estar involucradas en la diseminación de diversas enfermedades neoplásicas en seres humanos, contribuyendo activamente a la formación de metástasis51. Como mecanismo intermediario, es importante notar asimismo que el PDGF y el IGF-1 estimulan la proliferación celular y la fibrinogénesis por medio del estímulo inducido por el PDGF sobre el antiportador Na+/H+ y el consiguiente aumento del pHi, estímulo que a su vez es anulado por estimuladores del antiportador Na+/H+ 35. Si se tiene en cuenta que esta desviación patológica del equllibrio acidobásico en el sentido de una alcalinización celular es clave, específica y fundamental tanto en la transformación maligna como en la proliferación celular, habrá que tener en cuenta que cualquier mecanismo que tienda a producir una elevación patológica del pHi tiene muchas posibilidades de inducir la aparición y/o diseminación de un proceso maligno32, 47, 48.
Niveles plasmáticos elevados de VEGF son considerados un marcador de la actividad del proceso canceroso, tal como sucede en sarcomas de tejidos blandos en seres humanos, reflejando tanto el grado del tumor como la respuesta al tratamiento52. De forma similar, las tasas de bFGF en la orina de pacientes recientemente diagnosticados de leucemia están mucho más elevadas que en la normalidad —hasta siete veces— e incluso más elevadas que en cualquier otro proceso neoplásico39. Surgen entonces dudas adicionales de si la utilización exógena de un potente factor trófico como el VEGF y otros factores de crecimiento, mientras muestran un valor terapéutico potencial en la neurogénesis y la estimulación neuronal53, podrían representar un serio peligro como estimuladores de la oncogénesis y del proceso metastático. De ahí la racionalidad de que diversas terapias antitumorales modernas apunten a la inhibición del VEGF en un intento de controlar la angiogénesis tumoral50, 54, 55, o a la inkibición del PDGF con imatinib, etc.21. Hay que apuntar que en lo que se refiera al efecto de cambios acido-básicos sobre la apoptosis, la inducción de BFGF y la expresión de VEGF también existen publicaciones contradictorias al respecto31, 56.
Estos modernos hallazgos y aplicaciones implican la posibilidad teórica de que la aplicación local de factores de crecimiento, como arma terapéutica, puedan estar de alguna manera relacionada a la aparición de un cáncer de novo. Sin embargo, la posibilidad de que esto suceda in vivo es muy remota. Primero, el período estimulatorio de los GFs es relativamente corto23. En segundo lugar, no hay que confundir el hecho de que en los pacientes tratados no existe neoplasia alguna en la zona de tratamiento, luego no hay patología de este tipo a la que se pudiera influir negativamente. En tercer lugar, no tiene por qué existir ni la más mínima repercusión sistémica en cuanto a las tasas de factores de crecimiento secundarias a la aplicación local extravascular de concentrados de plaquetas.
De lo anterior se puede deducir que ni incluso la presencia de un tumor maligno en áreas del organismo alejadas de la localización tratada (articulación, músculo, cirugía maxilofacial, etc) debería considerarse a priori como una contraindicación terapéutica. Tal vez, por precaución, hay que considerar que en el momento actual, y a falta de más datos, debería evitarse la aplicación de concentrados de plaquetas en casos de cirugía maxilofacial o implantes bucales en personas que hayan presentado con anterioridad un tumor maligno de la esfera otorrinolaringológica. Finalmente, la larga experiencia clínica en cientos de casos a lo largo de varios años no ha dado lugar a la aparición de metaplasias o de ninguna otra lesión premaligna o maligna (E. Anitua, comunicación personal).
Conclusiones
Avanzando por el canto de la moneda
La pretensión de este trabajo no es la de ser una revisión en profundidad de los intricados caminos de señalización intracelular en la homeostasis y la apoptosis celular en diversas patologías, sino abrir nuevas vías conceptuales previamente no contempladas. La primera, y tal vez la principal conclusión, ha de ser que, a la vista de la literatura actual, y previa integración de los conceptos aquí desarrollados (Tabla I), se debe pensar que el fracaso de los actuales tratamientos, ya sea en oncología o en ENDs, puede tener lal misma raíz pero con consecuencias opuestas (imposibilidad de conseguir una apoptosis selectiva en cáncer y, por otra parte, imposibilidad de conseguir una efectiva antia~Fi poptosis en Alzheimer y otras ENDs.
En general, sólo a través de nuevas e inexploradas asociaciones de ideas puede llegarse a una visión global y omnicomprensiva de los problemas, para desde ahí poder incidir con mejores medios y bases de apoyo en la comprensión fisiopatológica de los procesos estudiados. Ya que sólo podemos curar lo que podemos comprender (Otto Warburg), visiones amplias y enfoques integrales de esta naturaleza pueden permitir avanzar en una mejora en el tratamiento basada en una comprensión más integral de estos procesos y de su etiopatogenia.
Por todo ello, se llega a las siguientes conclusiones en lo que se refiere a las relaciones entre las enfermedades degenerativas (ENDs), el cáncer y la aplicación de factores tróficos y de crecimiento en la clínica humana:
a) En cuanto a posibilidades de mejorar el tratamiento de las ENDs, y a la vista de los conocimientos y posibilidades actuales, se hace muy aconsejable la realización de estudios clínicos para detectar la presencia, disminución o incluso ausencia, de ciertos factores de crecimiento, tanto en el plasma humano (aspecto sistémico) como/o en las plaquetas y células del SNC u otras (aspecto celular)57 de enfermos con enfermedad de Alzheimer y otras ENDs. Esto supondría la posibilidad de detectar anormalidades cuantitativas y cualitativas, sistémicas o celulares, que puedan estar relacionadas, en una relación de causa a efecto, con la patogenia de ciertas enfermedades neurodegenerativas, y muy en especial con la etiopatogenia de la enfermedad de Alzhelmer. De ser así, y existen motivos suficientes para pensar en ello, se abriría una nueva línea encaminada al tratamiento de esta/s terrible/s enfermedad/es mediante la utilización, autóloga o heteróloga, de estos y otros factores de crecimiento en la clínica humana (VEGF, PDGFs, ésteres de forbol, tirosinoquinasas, proteinoquinasa C, activadores del antiportador Na+/H+, productos génicos tipo BCL-2, antioxidantes, etc. (Tabla I).
b) A la vista de la evidencia existente sobre la necesidad de evitar una acidificación intracelular para inhibir la apoptosis, un enfoque antiapoptótico basado en un entoque homeostático "ácido-básico" a nivel molecular abre nuevas vías terapéuticas destinadas al mantenimiento de una homeostasis celular dentro de límites fisiológicos58. Esto supone un intento de evitar la apoptosis mediada por, y a la vista de la literatura actual, una más que probable hiperacidificación intracelular espontánea en ENDs, y así en la prevención y tratamiento de las enfermedades degenerativas del sistema nervioso, más allá de la activación o inhibición de otros mecanismos mediados por factores diversos como los arriba señalados. Por lo anteriormente dicho, la utilización de todo factor estimulador del antiportador Na+/H+, tanto como la aplicación o estímulo de la producción endógena de ciertos factores de crecimiento y la prevención de una acidificación celular, estaría indicada en la prevención y tratamiento de las ENDs23.
c) Sin embargo, hay que tener presente que cualquier factor que estimule el antiportador Na+/H+, tanto como la aplicación de los PDGFs y otros factores contenidos en las plaquetas, podrían acelerar un proceso maligno localmente presente, e incluso inducirlo de novo. Sin embargo, en lo que se refiere a estas posibilidades, al menos al día de hoy, no existe evidencia alguna en la práctica médica que permita pensar que los procedimientos de aplicación local de factores de crecimiento dependientes de las plaquetas puedan constituir un peligro de degeneración neoplásica en la clínica humana ni influir en la progresión tumoral o la diseminación metastática en pacientes previamente no diagnosticados de un proceso maligno.
d) En cuanto a la aplicación de estos conceptos al tratamiento de las enfermedades neoplásicas, la lógica dicta que los esfuerzos deben de ir dirigidos a la inhibición de estimulación trófica, ya sea mediante la inhibición específica de determinados factores de crecimiento, y/o por acidificación celular, estimulación de la apoptosis tumoral36, 59-61 y/o antiangiogénesis39, 40, 43, 50, 55. Es este un tema que está siendo tratado ampliamente por una infinidad de publicaciones y grupos, algunos de los cuales son aquí citados, y lo que constituye de por sí una, tan prometedora como completa, área de investigación en la terapéutica de las enfermedades oncológicas en la actualidad.
Agradecimientos
El autor desea expresar su agradecimiento al Servicio Bibliotecario de la casa de la Cultura de Vitoria y al Departamento de Cultura de la Diputación de Alava, así como al Dr. Gorka Orive, por continua ayuda bibliográfica. De la misma forma le agradece al Dr. José Luis Arranz por su estímulo e inspiradoras sugerencias.
Bibliografía
1. Riedl AJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nature Revs Mol Cell Biol 2004; 5: 897-907. [ Links ]
2. Liu D, Martino G, Thangarolu M et al. Caspase-8-mediated intracellular acidification preceds mitochondrial dysfunction in somatostatin-induced apoptosis. J Biol Chem 2000; 275(13) 9244-50. [ Links ]
3. Rideout HJ, Stefonis L. Caspase inhibition: A potential strategy in neurological diseases. Histal Histopathol 2000; 16:895-908. [ Links ]
4. Waldelmer PC, Tatton W. Interrupting apoptosis in neurodegenerative disease: potential for effective therapy? Drug Discover Today (DDT) 2004; 9(5):210-8. [ Links ]
5. Waldeimer P. Prospects for antiapoptotic drug therapy of neurodegenerative diseases. Progress in Neuro Psychopharm & Biol Psychiatry 2003; 27:303-21. [ Links ]
6. Reed JC, Huang Z. Apoptosis pathways and drug targets. Nature Rev Drug Discov 2004; 3(111):895-981. [ Links ]
7. Reed, JC. Apoptosis-based therapies. Nat Rev Drug Discov 2002; 1:111-21. [ Links ]
8. Sadowski-Debling K, Coy JF, Mier W, Hug H, Los M. Caspases -their role in apoptosis and other physiological processes as revealed by knock-out studies. Arch Immunol & Therapiae Experimentalis 2002; 50:19-34. [ Links ]
9. Renz A, Berdel WE, Kreuter M, Bella C, Schulize-Osthoff K, Los M. Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood 2001; 98:1542-8. [ Links ]
10. Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inkibitors. Nature Revs Drug Discovery 2004; 3(12):1001-10. [ Links ]
11. Los M, Burek Ch, Stroh Ch, Benedyk K, Hug H, MacLiewitz A. Anticancer drugs of tomorrow: apoptotic pathways as targets for drug design. Drug Disc Today 2003; 8:67-77. [ Links ]
12. Resnick L, Fennell M. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Disc Today 2004; 9: 932-9. [ Links ]
13. Khaled AR, Moor AN, Li A et al. Thropic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1 which induces intracellular alkalinization. Mol & Cell Biol 2001; 21:7545-57. [ Links ]
14. Tatton WG, Chamblers-Redman R, Ju WJH et al. Propargylamines induce antiapoptotic new protein synthasis in serum- and nerve growth factor (NGF)-withdrawn, NGF-differentiated PC-12 cells. J Pharm & Exp Therap 2002; 301: 753-64. [ Links ]
15. Nuydens R. Dispersyn G. Van Den KieLoom G, M. et al. Bc1-2 protects neuronal cells aguinst taxol-induced apoptosis by inducing multi-nucleation. Apoptosis 2000; 5: 335-43. [ Links ]
16. Harguindey S, PeUraz J L, García Cañero R, Katin M. Edelfosine, apoptosis, MDR and Na+/H+ exchanger: induction mechanisms and treatment implications. Apoptosis 2000; 5:87-9. [ Links ]
17. DiGiammarino J, Lee ADS, Cadwell C et al. A novel mechanism of tumorogenesis involving pH-dependent desestabilization of a mutant p53 tetramer, Nature Struct Biol 2000; 9(1):12-6. [ Links ]
18. Harguindey S, Orive G, Pedraz JL. Integración multidisciplinaria de campos oncológicos a través de mecanismos cruciales y vías comunes. De etiopatogénesis a tratamiento. Oncología 2002; 25(7):295-316. [ Links ]
19. Williams AC, Collard TJ, ParasEeva C. An acidic emironment leaUs to p53 dependent induction of apoptosis. Oncogene 1999; 18:3199-3204. [ Links ]
20. Tatton W, Chen D, Chamblers-ReUman R, Wheeler L, Nixon R, Tatton N. Survey Ophtalmology 2003; 48 (suppl 1): s25-s37. [ Links ]
21. López Martín A, Colomer Bosch R. Receptores de c-Kit y del factor de crecimiento derivado de las plaquetas como diana para el tratamiento de tumores sólidos. Rev Oncol 2003; 5(Supl):13-21. [ Links ]
22. Marches R, Vitetta ES, Uhr JW. A role for intracellular pH in membrane IgM-mediated cell death of human B Iymphomas. PNAS 2001; 98:3434-9. [ Links ]
23. Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous plateléts as a source of proteins for healing and tisuue regeneration. Thromb Haemost 2004; 91:415. [ Links ]
24. Matsuyama S, Llopis J, Deveroux QL, Tsien RY, Reed JC. Changes in intramitchondrial and cytosolic pH: early events tRat modulate caspase activation during apoptosis. Nature Cell Biol 2000; 2:18-35. [ Links ]
25. Li J, Eastman A. Apoptosis in an interleuEin-2-dependent cytotoxic T IympoLocyte cell line is associated wilh intracellular acidification. Role of the Na+/H+-antiport. J Biol Chem 1995; 270(2):3203-11. [ Links ]
26. Hamilton G, Cosentini EP, Teleky B, et al. The multidrugresistance modifiers verapamil cyclosporine A and tamoxifen induce an intracellular acidification in colon carcinoma cell lines in vitro. Anticancer Res 1993; 13(6A):2059-63. [ Links ]
27. Gottlleb RA, Norberg J, Skowronski E, Babior BM. Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. PNAS 1996; 93:654-8. [ Links ]
28. Rebollo A, Gómez J, de Aragón AM, Lastres P, Silva A, Pérez-Sala D. Apoptosis induced by IL-2 withdrawal is associated with an intracellular acidification. Exp Cell Res 1995; 218:581-5. [ Links ]
29. Thangaroju M, Sharma K, Leber B, Andrews DW, Shen S-H, SriLant B. Regulation of acidification and apoptosis by SHP-1 and BCL-2. J Biol Chem 1999, 274: 29549-57. [ Links ]
30. Park HJ, Lyons JC, Ohtsubo T, Song CW. Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer 1999; 80(2):1892-7. [ Links ]
31. Lagadic-Gossman D, Huc L, Lecureur V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death & Diherentiation 2004; 11:953-61. [ Links ]
32. Harguindey S, Pedraz JL, García Cañero R, Pérez de Diego J, Cragoe EJ Jr. Hydrogen ion-dependent oncogenesis and parallel new avenues to cancer prevention and treatment using a H+-mediated unifying approach: pH-related and pH-unrelated mechanisms. Critical Rev Oncogenesis 1995; 6(1):1-33. [ Links ]
33. Roepe PD, Wei LY, Cruz J, Carlson D. Lower electrical membrane and altered pHi homeostasis in multidrugresistant (MDR) cells: further characterization of a series of MDR cell lines expressing different levels of Pglycoprotein. Biochamistry 1993; 32:11042-56. [ Links ]
34. Simon S. The multiple mechanisms of drug resistance and cellular pH. En: Gillies R, ed. The tumor microenvironment: causes and consequences of hypoxia and acidity. Novartis Foundotion Symposium. Chichester-New York: John Wiley & Sons, Ltd., 2001; 240:269-81. [ Links ]
35. Di Sario A, Baroni GS, Bendia E et al. Intracellular pH regulation and Na+/H+ exchange activity in human hepatic stellate cells: effect of platelet-derived growth factor, insulin-like growth factor 1 and insulin. J Hepatol 2001; 34(3):378-85. [ Links ]
36. Rich IR, WortLinglon-White, OA, Musk P. Apoptosis of leukemic cells accompanies reduction in intracellular pH aker targeted inFibition of the Na+/H+ exchanger. Blood 2000; 95:1427-34. [ Links ]
37. Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature Revs Cancer 2004; 4:937-47. [ Links ]
38. Sánchez-Martín M, Sánchez-García I. Inhibidores de BCR-ABL como un abordaje molecular en la leucemia mieloide crónica. Rev Oncol 2003; 5(Supl):4-12. [ Links ]
39. Folkman J, Browder T, Palmblad J. Angiogenesis research: guidelines for translation to clinical applications. Thromb Haemost 2001; 86:23-33. [ Links ]
40. Harguindey S. Use of Na+/H+ antiporter inhibitors as a novel approach to cancer treatment. Cragoe EJ Jr, Kleyman ThR, Simchowitz L, eds. Amiloride and its analogs: unique cation transport inkibitors. New York: VCH Publishers Inc,1992; 317-34. [ Links ]
41. Gatenby RA, Gawlinski T. The glycolytic phenotype in carcinogenesis and tumor invasion: insights through mathematical models. Cancer Res 2003; 63:3847-54. [ Links ]
42. Braña MF, Domínguez G, Añorbe L. Angiogenesis and cancer: the contribution of organic chemistry. Rev Oncol 1999; 1:122-32. [ Links ]
43. Izumi HT, Torigoe T, Ishiguchi H et al. Cellular pH regulators: potentially promising molecular targets of cancer chematherapy, Cancer Treat Rev. 2003; 29:541-9. [ Links ]
44. Reshkin SJ, Bellizzi A. Cardone M et al. Paclitaxel induces apoptosis via Protein Kinase A- and p38 Mitogenactivated Protein-dependent inkibition of the Na+/H+ Exchanger (NHE) Isoform 1 in Human Breast cancer celis, Clinical Cancer Res 2003; 9:2366-73. [ Links ]
45. Harguindey S. Integrating fields of cancer research through pivotal mechanisms and synthetic final pathways: A unifying and creative overview. Med Hypotheses 2002; 58(6):444-52 -plus "Erratum": Med Hypoth 2003; 61 (2): 318-19. [ Links ]
46. Chen Q, Benson RSP, Whetton AD et al. Role of acidbase homeostasis in the suppression of apoptosis in haemopoyetic cells by v-abl tyrosine kinase. J Cell Sci 1997; 110: 379-87. [ Links ]
47. Reshkin SJ, Bellizzi A, Caldeira S et al. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformationassociated pLenotypes, FASEB J 2000; 14:2185-97. [ Links ]
48. Perona R, Serrano R. Increased pH and tumorigenicity of fibroLlasts expressing a yeast proton pump. Nature 1988; 334:438-40. [ Links ]
49. Reynolds JE, Li JF, Craig RW, Eastman A. BCL-2 and MCL-1 expression in chinese hamster ovary cells inkibits intracellular acidification and apoptosis induced by staurosporine. Exp Cell Res 1996; 225:430-6. [ Links ]
50. Orive G, Reshkin SJ, Harquindey S, Pedraz JL. Hydrogen ion dynamics and the Na+/H+ antiporter in cancer angiogenesis and antiangiogenesis. Br J Cancer 2003; 89:395-9. [ Links ]
51. Pinedo HM, Verheul, D'Amato RJ, Folkman J. Involve- ment of platelets in tumor angiogenesis? Lancet 1998; 352: 1775-7. [ Links ]
52. Hayes AJ, Mostyn-Jones, Koban MU, A Hern R, Burton P, Thamas JM. Serum vascular endothelial growth factor as a tumor marker in soh tissue sarcoma. Br J Surg 2004; 91 (2):242-7. [ Links ]
53. Cao L, Jico X, Zuzga DS et al. VEGF links hippocampal activity with neurogenesis, learning and memory. Nature Genetics 2004; 36:827-35. [ Links ]
54. Gordon MS. Novel antiangiogenic therapies for renal cancer. Clin Cancer Res 2004; 10(18 Pt 2): 6377S-6381S. [ Links ]
55. Harguindey S, Orive, G, Pedraz JL, Bello G, Arranz JL, Samaniego JM. Curación aparente de un caso de carcinoma de ovario metastático después de tratamiento crónico con inhibidores del antiportador Na+/H+. Oncología 2002; 25: 472-6. [ Links ]
56. D'Arcangelo D, Facchiano F, Barluchi, LM et al. Acidosis inhibits endothelial cell apoptosis and function and induces basic fibroblast growth factor and vascular endothelial growth factor expression. Circ Res 2000; 86:312-8. [ Links ]
57. Aguayo A, Estey E, Kantarjian H et al. Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute leukemia. Blood 1999; 94:3717-21. [ Links ]
58. Pérez-Sala D, Collado-Escobar D, Mollinedo F. Intracellular alkalinization supresses lovastatin-induced apoptosis in HL60 cells through the inactivation of a pH-dependent endonuclease. J Biol Chem 1995; 270:6235-42. [ Links ]
59. Li J, Eastman A. Apoptosis in an interleukin-2-dependent cytotoxic T Iymphocyte cell line is associated with intracellular acidification. Role of the Na+/H+-Antiport. J Bial Chem 1995; 270:3203-11. [ Links ]
60. Wolf CM, Reynolds JE, Morana SJ, Eastman A. The temporal relationship between protein phosphatase. ICE/CED-3 proteases, intracellular acidification, and DNA fragmentation in apoptosis. Exp Cell Res 1997; 230(1):22-7. [ Links ]
61. Angoli D, Delia D, Wank E. Early cytoplasmatic acidification in retinamide-mediated apoptosis of human leukemia cells. Biochem Biophys Res Com 1996; 229(2):681-5. [ Links ]
 Dirección para correspondencia
Dirección para correspondencia
Dr. S. Harguindey
C/ Postas, 13, 3° izq
E-01004 Vitoria (Alava)
E-mail: salvaszh@telefonica.net
Recibido: 14.12.04
Aceptado: 20.12.04

