










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkOncología (Barcelona)
versión impresa ISSN 0378-4835
Oncología (Barc.) vol.29 no.4 abr. 2006
NOTA CLÍNICA
Tumor de colisión renal: un caso inusual de carcinoma de células renales y leiomiosarcoma renal
V. Conde; J. R. Delgado; E. González; R. Luque; C. Rodríguez; C. M. Sánchez; B. Jiménez; A. Montesa; A. Irigoyen; P. Ballesteros
Servicio de Oncología Médica
Hospital Universitario Virgen de las Nieves
Granada (España)
RESUMEN
PROPÓSITO: En el tumor de colisión coexisten dos tumores histológicamente diferentes sin mezcla en la interfase. Los tumores de colisión que afectan al riñón son extremadamente raros.
MATERIAL Y MÉTODOS: Presentamos el caso de una paciente con un tumor de colisión renal formado por un carcinoma de células renales y un leiomiosarcoma renal.
RESULTADOS: El manejo clínico de este caso fue complejo debido a la escasa experiencia en el tratamiento de este tipo de tumores y los pobres resultados que se obtienen con el tratamiento sistémico en ambos tipos de tumores.
CONCLUSIONES: Existen muy pocas comunicaciones sobre tumores de colisión renales. Consideramos de interés el conocimiento sobre la existencia de estos tumores así como su complejidad clínica.
Palabras clave: Tumor de Colisión. Carcinoma de células renales. Leiomiosarcoma renal.
SUMMARY
PURPOSE: A collition tumor is characterized by the coexistence of two histologically different neoplasms that are independent of any interface mixing. Kidney collition tumors are extremely rare.
MATERIAL AND METHODS: We present the case of a patient diagnosed of a kidney collition tumor consisting of renal cell carcinoma and renal leiomyosarcoma.
RESULTS: The clinical management of this patient was complex due to the limited experience in the treatment of his kind of tumors, and to the poor results obtained with systemic treatment of the two types of tumors involved.
CONCLUSIONS: There are very few communications about collition tumors of the kidney, and we consider interesting to repot the existence of these tumors as well as their clinical complexity.
Key words: Collition tumor. Kidney. Renal cell carcinoma. Renal leiomyosarcoma.
Introducción
El tumor de colisión es una rara entidad histopatológica en la que coexisten dos tumores histológicamente diferentes sin mezcla en la interfase1. Hay determinados tumores de colisión que ocurren de forma relativamente más frecuente, como por ejemplo el colangiocarcinoma junto con el hepatocarcinoma, pero los tumores de colisión que afectan al riñón son extremadamente raros1.
El cáncer de células renales constituye el 3% de los tumores sólidos que aparecen en el adulto2-4, siendo el subtipo histológico carcinoma de células claras el más frecuente (75%) seguido del tipo papilar (15%)2. Tan solo un 3% de los tumores que aparecen a nivel renal son sarcomas4-6, siendo el leiomiosarcoma el subtipo histológico que con más frecuencia aparece5-8, constituyendo tan solo del 0,5 al 1,5% de todos los tumores malignos renales7.
En este artículo presentamos el caso de un tumor de colisión renal constituído por un cáncer de células renales tipo papilar y un leiomiosarcoma renal, teniendo como objetivo fundamental destacar la excepcionalidad del caso (no se ha encontrado ningún caso semejante en la revisión de la literatura realizada) y como objetivo secundario poner de manifiesto la dificultad en el manejo diagnóstico-terapeútico de este caso en concreto.
Caso clínico
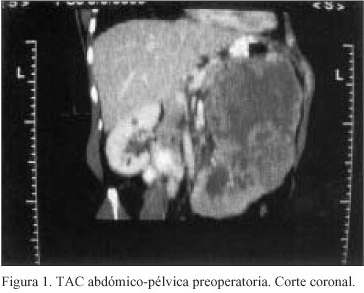
Paciente de 53 años de edad en el momento del diagnóstico y antecedentes personales de una cesárea, hipertensión arterial y taquicardia en tratamiento con Atenolol, que acude al Servicio de Urgencias por un cuadro de 5 meses de evolución de dolor sordo en flanco izquierdo de carácter progresivo y microhematuria. Se realizan ecografía y posteriormente tomografía axial computerizada (TAC) abdómino-pélvica que evidencian una gran masa sólida de 20x16x13 cm que surge del polo renal izquierdo, sin trombosis venosa asociada (Figuras 1 y 2). Se observan a su vez adenopatías para-aórticas. Todo ello compatible con tumor renal izquierdo.

Ante este hallazgo se realiza estudio de extensión con analítica general en la que se detecta anemia microcítica ferropénica e hiperuricemia leves, una radiografía de tórax que es normal, una gammagrafía ósea en la que se observa una imagen con leve aumento de captación del trazador correspondiente a partes blandas, de morfología redondeada, localizada en hipocondrio y vacío izquierdos rebasando la línea media y 3 focos de hipercaptación del trazador uno en pala iliaca izquierda, el 2º en hemivértebras izquierdas L4 y L5 y el 3º a la altura de hemivértebra derecha L2 que podrían corresponderse con afectación ósea por contigüidad o ser de carácter metastásico y una resonancia magnética abdominal que confirma los hallazgos de la TAC.
Con el diagnóstico de presunción de carcinoma de células renales con adenopatías retroperitoneales la paciente es sometida a nefrectomía radical izquierda, extirpación de toda la grasa pararrenal y de la glándula suprarrenal izquierda y linfadenectomía paraaórtica e interaortocava desde la arteria renal hasta 3 cm de la iliaca común. La extirpación de la masa resultó dificultosa por infiltración de mesocolon transverso y cola de páncreas que se liberan quedando respetado el mesocolon. El resultado anatomo-patológico (A.P.) fue de: Tumor renal de colisión (Leiomiosarcoma (21 x15 cm) y Carcinoma renal tipo papilar grado nuclear 3 (7 x 3,5 cm)). El peso de la pieza en su conjunto fue de 2539 gramos. (Figuras 3 y 4). La tumoración contactaba con el borde quirúrgico en la mayoría de las zonas. El parénquima renal estaba respetado microscópicamente y no se observaba infiltración tumoral. El fragmento ureteral y el hilio renal se encontraban libres de infiltración tumoral. El perfil inmunofenotípico que presentaba el tumor fue el siguiente: Actina, Desmina, S-100, Sinaptofusina y CD 56 y c-kit negativos; Actina de músculo liso positiva en la zona sarcomatosa y cóctel de queratina (E1, E3) positivo en la zona carcinomatosa. A nivel de la cadena para-aórtica se aislaron 16 adenopatías, la mayor de 2,5 cm, presentando metástasis 14 de ellas, 13 del carcinoma y 1 con metástasis mixta (sarcoma+carcinoma). En la cadena interaortocava se aislaron 6 adenopatías, la mayor de 1,4 cm, siendo metástasis del carcinoma 3 de ellas. El lecho mesocólico se encontraba infiltrado por leiomiosarcoma. En el tejido adiposo perisuprarrenal se aislaron 4 adenopatías, 3 de ellas con metástasis del carcinoma y otra con metástasis mixta (carcinoma+sarcoma). La glándula suprarrenal izquierda, la grasa perirrenal y la vesícula no presentaban elementos tumorales.


Nos encontramos pues ante un tumor de colisión renal constituído por un cáncer renal tipo papilar estadio IV (pT3-4pN2) según la clasificación TNM y un leiomiosarcoma renal estadio IV (pT2bpN1) según la clasificación de la AJCC no resecado de forma radical y con posibles metástasis óseas por gammagrafía ósea.
El postoperatorio cursa sin complicaciones y la paciente es remitida al Servicio de Oncología Médica. Se decide proponer tratamiento complementario con quimioterapia según esquema Ifosfamida 5 g/m2 en infusión contínua de 24h x 1día + Adriamicina 60 mg/m2 x 1 día/21 días frente al componente sarcomatoso del tumor. Previamente se solicita TAC en la que se observa pequeño aumento de partes blandas por detrás de la cola pancreática y lecho renal que podría ser compatible con enfermedad presente.
La paciente inicia tratamiento según el esquema previsto 3 semanas después de la cirugía. Recibe un total de 6 ciclos con buena tolerancia clínica. Tras el 4º ciclo se realiza una TAC abdominal que es normal, y al finalizar el 6º ciclo se realiza gammagrafía ósea que no presenta hallazgos patológicos. La paciente pasa a revisiones periódicas y 3 meses más tarde se realiza una radiografía de tórax en la que aparecen imágenes sugerentes de metástasis pulmonares bilaterales que se confirman por TAC en la que se observan múltiples metástasis pulmonares bilaterales milimétricas (Figura 5). Con el fin de filiar el origen de dichas metástasis se consulta con Cirugía Torácica y se decide realizar videotoracoscopia izquierda con toma de biopsias. El diagnóstico A.P. fue de metástasis de Carcinoma pobremente diferenciado con componente epitelial (renal). Ante este diagnóstico 6 meses después de finalizar el primer esquema de quimioterapia se decide iniciar una segunda línea de tratamiento con un esquema de quimioterapia con Gemzitabina y Fluoropirimidinas9 que se ha mostrado útil en el carcinoma renal estadio IV: Gemcitabina 1000 mg/m2 días 1 y 8 + Capecitabina 1000 mg/m2/12h días 1-14/21 días, que la paciente acepta.

Se inicia tratamiento con una reducción de dosis del 20% que se mantiene el resto del tratamiento dado el estado general (ECOG: 1-2) de la paciente. Tras el 2º ciclo la paciente sufre como complicación un tromboembolismo pulmonar del que se recupera pero que provoca un retraso de 4 semanas en la administración del 3º ciclo. Tras 6 ciclos de tratamiento que recibe con aceptable tolerancia salvo anemia grado 4 se realiza reevaluación con TAC corporal en la que se evidencia persistencia de las metástasis pulmonares con aparición de metástasis hepáticas, esplénicas y recaída local.
Ante esta progresión se propone tratamiento con IL-2 durante 6 semanas (1 semana de inducción con 18 Millones de Unidades (MU) x 5 días y 5 semanas: 9 MU días 1 y 2 y 18 MU días 3 a 5)10. La paciente acepta el tratamiento que recibe con una toxicidad moderada con cuadro constitucional secundario grado 2, anemia grado 3 y emesis grado 1, manteniendo el estado general.
Al finalizar el tratamiento se realiza nueva reevaluación con TAC que evidencia progresión de la enfermedad con gran masa en el lecho quirúrgico de 19x10x5cm, que había crecido con respecto a la TAC anterior, y persistencia de las metástasis en el resto de las localizaciones previas. La paciente muestra empeoramiento del estado general con aparición de dolor abdominal y lumbar y en la exploración física se palpa masa epigástrica de 5 cm correspondiente con la masa subyacente.
Ante esta nueva progresión se considera que el tumor es resistente a esquemas de quimio o inmunoterapia frente al carcinoma renal y se propone iniciar tratamiento paliativo con Adriamicina liposomial frente al componente sarcomatoso del tumor. La paciente acepta la propuesta y recibe un primer ciclo. Sin embargo a la semana acude a Urgencias por cuadro de shock hipovolémico con acidosis metabólica e insuficiencia renal prerrenal secundario a hiperemesis de 4 días de evolución y anemia grado 4. La paciente se recupera de este episodio pero a la semana comienza con vómitos fecaloideos de probable origen obstructivo por compresión de la masa retroperitoneal, produciéndose deterioro progresivo de la paciente y la paciente fallece por fracaso multiorgánico a los 19 meses del diagnóstico.
Discusión
Como ya se ha comentado los tumores de colisión a nivel renal son extremadamente raros. En el caso anteriormente descrito se da la circunstancia de estar formado por dos tumores a su vez muy poco frecuentes dentro del conjunto de los tumores sólidos que pueden aparecer en los adultos: por un lado un cáncer de células renales que constituye el 3% de los tumores sólidos del adulto y por otro un sarcoma de partes blandas cuya incidencia es menor del 1% de todas las neoplasias11, localizándose éste en un 34% de los casos a nivel intraabdominal: un 15% de los casos aparecen a nivel retroperitoneal y un 19% a nivel visceral8. El leiomiosarcoma es el subtipo histológico que con más frecuencia aparece a nivel renal5-8 y aunque el leiomiosarcoma renal en ocasiones puede confundirse con un cáncer de células renales con diferenciación sarcomatoide las técnicas de inmunohistoquímica nos permiten diferenciar perfectamente estos dos tipos de tumores5.
La supervivencia global a los 5 años del cáncer de células renales cuando está localizado es del 50 al 60%3. Cuando se trata de tumores metastásicos la supervivencia baja dramáticamente, cursando en algún caso de forma indolente durante años. El cáncer de células renales es uno de los pocos tumores que puede presentar remisiones espontáneas sin tratamientos activos, pero esto es raro y no conduce a largas supervivencias3. Por otra parte la mortalidad global de los sarcomas de partes blandas es del 50%11 y la curación es rara cuando se diagnostican en fase localmente avanzada. La mediana de supervivencia de los sarcomas de partes blandas metastáticos es de 8-12 meses, aunque un 20-25% de pacientes viven más de 2 años8.
Desconocemos cuál puede ser el pronóstico de un tumor de colisión y en concreto uno de las características que hemos descrito. Por tanto nos parece razonable analizar los factores pronósticos de cada tumor de forma independiente y en función de los mismos tomar las decisiones terapeúticas.
Los principales factores pronósticos del cáncer de células renales son: el tamaño, la presencia de metástasis ganglionares y a distancia, el grado histológico tumoral, la extensión a órganos adyacentes, la afectación de la vena cava3 y el subtipo histológico (por ejemplo el cáncer tipo papilar tiene mejor pronóstico que el cáncer de células claras)2. Sin embargo y aunque siempre se ha considerado la afectación de la vena renal como un factor de mal pronóstico su repercusión en la supervivencia no parece ser tan marcado como se suponía2. En la enfermedad metastásica los factores pronósticos de supervivencia son: el intervalo libre de enfermedad, la nefrectomía previa, la localización y número de las metástasis y el estado general del paciente3.
Los factores de mal pronóstico del sarcoma de partes blandas son: el tamaño superior a 5 cm, el alto grado histológico de malignidad, la presencia de márgenes positivos tras la cirugía, la localización profunda, la recaída local y la presencia de metástasis8, 11. Otros factores de mal pronóstico que se han presentado en algunas series son: la edad, la infiltración de nervios periféricos y el subtipo histológico (el fibrosarcoma aumentaría el riesgo de recaída local y el leiomiosarcoma el riesgo de recaída a distancia)12. Estos factores pronósticos son perfectamente aplicables a los leiomiosarcomas renales, aunque en general estos tumores, tal y como refleja la literatura suelen ser tumores de especial mal pronóstico, con tendencia a la recaída local y a distancia4-6, presentando supervivencias inferiores en la mayoría de los casos a los dos años4, 5.
La base del tratamiento del cáncer de células renales en estadios localizados es la nefrectomía radical2, 3. El papel de la linfadenectomía resulta aún controvertido3. En pacientes con ganglios positivos no existe evidencia de que el tratamiento adyuvante con inmunoterapia (IL-2 o IFN-a) aumente la supervivencia2. El cáncer renal metastásico tras diversos estudios se ha considerado clásicamente quimio y hormono-resistente2, 3, sin embargo algunos estudios con poliquimioterapia basados en Fluoropirimidinas y Gemzitabina parecen presentar una mayor tasa de respuestas2. La terapia anti-angiogénica también se ha mostrado eficaz en este tipo de tumores13. Sin embargo el tratamiento sistémico más aceptado del cáncer de células renales metastásico es la inmunoterapia, bien IL-2 o bien IFN-a, que son los únicos tratamientos que han sido capaces de inducir un mayor número de respuestas, algunas de ellas completas y duraderas2, 3, si bien la dosis y vía de administración del IFN-a aún no están bien perfiladas2. La combinación de estos agentes entre sí, así como con agentes quimioterápicos no han demostrado un aumento de la tasa de respuestas ni de la supervivencia2, 3.
El tratamiento de los sarcomas de partes blandas localizados se basa en la cirugía radical con márgenes de seguridad y linfadenectomía en el caso de presencia de ganglios positivos8, 11. El tratamiento complementario con radioterapia estaría indicado cuando la cirugía no ha podido ser radical y cuando se trata de tumores que presenta un grado histológico intermedio o alto11. El problema radica cuando la localización del tumor es retroperitoneal, como es nuestro caso, en el que la morbilidad que produce la radioterapia es importante y en muchas ocasiones inaceptable dados los grandes volúmenes que habitualmente hay que irradiar8, 11. El tratamiento adyuvante con quimioterapia está en discusión dada la heterogeneidad y bajo número de pacientes que incluyen los ensayos que se han realizado sin que éstos muestren un beneficio claro8, 11, pero se puede ofrecer a pacientes con un estadio III o ganglios positivos, ya que la supervivencia esperable a los 5 años de este tipo de pacientes es del 35% y el esquema de tratamiento debería incluír según algunos autores adriamicina e ifosfamida a altas dosis11. En el caso de la enfermedad metastásica, que presenta una mediana de supervivencia de 8 a 12 meses8 (aunque en algunos casos la supervivencia puede alcanzar los 2 años)8, 11, el manejo se basa en combinar la resección de las metástasis o de la recurrencia local siempre que sea posible y el tratamiento con quimioterapia cuya base continúan siendo las antraciclinas clásicas y liposomiales, así como la ifosfamida a altas dosis, en monoterapia o combinación8, 11. Sin embargo, en diferentes estudios los leiomiosarcomas de origen gastrointestinal se han mostrado especialmente resistentes a la doxorrubicina y a la ifosfamida8, 11. El manejo terapeútico de los leiomiosarcomas de origen renal no difiere del resto de los sarcomas de partes blandas5, 6.
En el caso clínico que presentamos nos encontramos con dos tumores "sincrónicos" a nivel renal, uno de ellos altamente infrecuente en dicha localización como es el leiomiosarcoma, con factores de muy mal pronóstico por su avanzado estadio, histología en el caso del leiomiosarcoma, y por la imposibilidad de una cirugía radical de inicio así como de radioterapia complementaria que hubieran ayudado al control local. Por otra parte el manejo terapéutico resultaba muy complejo desde el inicio, por lo avanzado de la enfermedad al diagnóstico, así como por otros factores como: la presencia de dos tumores con alto riesgo de recaída tanto local como a distancia y la ausencia de un tratamiento sistémico claramente efectivo tanto por el tipo histológico de ambos tumores como por la práctica inexistencia de ensayos clínicos randomizados y bien diseñados, dada la baja frecuencia de estos tumores.
Bibliografía
1. Wang BY, Strauchen JA, Rabinowitz D, Tillem SM, Unger PD. Renal cell carcinoma with intravascular lymphomatosis: A case report of an inusual collision tumors with review of the literature. Arch Pathol Lab Med. 2001; 125: 1239-1241. [ Links ]
2. Marston Linehan W, Bates SE, Yang JC. Cancer of the Kidney. En: De Vita VT, Jr, Hellman S, Rosenberg SA. Cancer: Principles and Practice of Oncology 7th Edition. Philadelphia: Lippincott Williams & Wilkins, 2005; 1139-1168. [ Links ]
3. Guillem V, Iborra I, Burriel C. Cáncer de riñón. En: Cortés-Funes H, Díaz-Rubio E, García-Conde J, Germà Lluch JR, Guillem Porta V, López López JJ, Moreno Nogueira JA, Pérez Manga G. Oncología Médica, Vol. 1. Madrid: Nova Sidonia Oncología-GRUPO AULA MÉDICA S.A., 1999; 699-718. [ Links ]
4. Rakowsky E, Barzilay J, Schujman E, Servadio C. Leiomyosarcoma of kidney. Urology 1987; 39(1): 68-70. [ Links ]
5. Davis R, Vaccaro JA, Hodges GF, Belville WD, Kiesling V, Jr. Renal leiomyosarcoma: Plea for aggressive therapy. Urology 1992; 40(2): 168-171. [ Links ]
6. Barros JM, Castro AM, Rodríguez B y col. Leiomiosarcoma renal. Actas Urol Esp. 1993; 17(3): 199-201. [ Links ]
7. Moazzan M, Ather MH, Hussainy AS. Leiomyosarcoma presenting as a spontaneously rupture: a tumor-case report. BMC Urol. 2002; 2: 13. [ Links ]
8. Brennan MF, Singer S, Maki RG, O'Sullivan B. Soft Tissue Sarcoma. En: De Vita VT, Jr, Hellman S, Rosenberg SA. Cancer: Principles and Practice of Oncology 7th Edition. Philadelphia: Lippincott Williams & Wilkins, 2005: 1581- 1637. [ Links ]
9. Rini Brian I, Vogelzang Nicholas J, Dumas Mary C, Wade James L, Taber David A, Stadler Walter M. Phase II trial of weekly intravenous Gemcitabine with continuous infusion Fluoracil in patients with metastatic renal cell cancer. J Clin Oncol. 2000; 18(12): 2419-2426. [ Links ]
10. Sleijfer DT, Janssen RA, Buter J, de Vries EG, Willemse PH, Mulder NH. Phase II study of subcutaneous interleukin-2 in unselected patients with advanced renal cell cancer on an outpatients basis. J Clin Oncol. 1992; 10(7): 1119-1123. [ Links ]
11. Poveda A, Querejeta A, Buesa JMª. Sarcoma de partes blandas. En: Cortés-Funes H, Díaz-Rubio E, García-Conde J, Germà Lluch JR, Guillem Porta V, López López JJ, Moreno Nogueira JA, Pérez Manga G. Oncología Médica, Vol. 2. Madrid: Nova Sidonia Oncología-GRUPO AULA MÉDICA S.A., 1999: 1041-1060. [ Links ]
12. Pisters P, Leung D, Woodruff J, et al. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol. 1996; 14(5): 1679-1689. [ Links ]
13. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of Bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003; 349(5): 427-434. [ Links ]
 Correspondencia:
Correspondencia:
Dr. J. R. Delgado Pérez
Jefe de la Unidad de Oncología Médica
Hospital de Día de Oncología Médica
Hospital Universitario Virgen de las Nieves
Avenida de las Fuerzas Armadas, s/n
E-18014 Granada (España)
Recibido: 10.02.06
Revisado. 13.02.06
Aceptado: 27.02.06

