










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkOncología (Barcelona)
versión impresa ISSN 0378-4835
Oncología (Barc.) vol.29 no.7 sep. 2006
REVISIÓN
Efectos de la radiación ultravioleta (UV) en la inducción de mutaciones de p53 en tumores de piel
C. M. Cabrera MoralesI; M. A. López-NevotII
Hospital Universitario Virgen de las Nieves; Granada (España)
IServicio de Anatomía Patológica
IIServicio de Análisis Clínicos
RESUMEN
El cáncer de piel representa el tipo mas frecuente de cáncer humano, con una incidencia que va en aumento en los últimos años. La exposición excesiva a la radiación ultravioleta (UV) de la luz solar se asocia de forma directa con la aparición de tumores no-melanoma; sin embargo esta asociación no es tan aparente en el melanoma. En los tumores de piel no-melanoma, la radiación ultravioleta es la responsable de la producción de mutaciones puntuales en genes relevantes como p53, consistentes en transiciones que ocurren en sitios dipirimidina. Encontrándose la acumulación de mutaciones en el gen supresor de tumores p53 inducidas por la radiación UV en el inicio del proceso tumoral de piel.
Palabras clave: Cáncer de piel. Radiación ultravioleta. p53.
SUMMARY
Skin cancer is presently the most frequent type of human cancer, showing in addition an increasing incidence in the last years. The excessive exposure to the sunlight ultraviolet (UV) radiation is associated with non-melanoma skin cancers, while such an association is not so apparent in melanoma. The hallmark of UV radiation in non-melanoma skin tumors is the high frequency of transition mutations at dipyrimidine sequences of relevant genes as p53. The accumulation of UV-induced p53 mutations seems to be critical in the development of skin cancer.
Key words: Skin cancer. Ultraviolet radiation. p53.
Introducción
Los tumores de piel representan el tipo más frecuente de neoplasias humanas. Prácticamente el 99% de ellos corresponden a tumores de piel no-melanoma, carcinoma basocelular (CBC) y espinocelular (CEC)1; el melanoma maligno representa un porcentaje muy pequeño, que sin embargo dada su agresividad es el responsable de la mayoría de los fallecimientos ocasionados por cáncer de piel2.
El carcinoma basocelular representa la forma más frecuente de cáncer de piel, seguido por el espinocelular3. Aunque son las formas más comunes de cáncer de piel, constituyen menos del 0.1% de las muertes producidas por cáncer. Ambos tipos suelen aparecer en individuos de complexión delgada que se han expuesto repetidamente a la radiación ultravioleta de la luz solar, y suelen aparecer con mayor frecuencia en los países del hemisferio sur4. Ambos tumores son de origen epitelial, el carcinoma basocelular se origina a partir de las células epidérmicas pluripotenciales de la capa basal y menos frecuentemente de los anexos cutáneos5. En algunos síndromes genéticos, y también en pacientes inmunodeprimidos, existe una especial predisposición a desarrollar carcinoma basocelular. Aproximadamente el 80% se desarrolla en sitios expuestos de cabeza y cuello. Es un tumor de lento crecimiento y las metástasis son excepcionales. El carcinoma espinocelular se origina a partir de los queratinocitos o sus anexos epidérmicos6. La epidemiología es similar al carcinoma basocelular en cuanto a edad de presentación, tipo de piel y exposición solar crónica. Los pacientes inmunodeprimidos tienen un riesgo aumentado de desarrollar el carcinoma espinocelular, existiendo evidencia de que en algunos casos se asocia con la infección por HPV7. Una forma precursora potencial del carcinoma espinocelular en un 5% de los casos es la queratosis actínica8. El melanoma maligno, sin embargo, se origina a partir de los melanocitos diseminados de las capas basales de la epidermis2. Sus finas prolongaciones citoplasmáticas se ramifican entre los queratinocitos hacia la superficie cutánea. Los melanocitos son responsables de la síntesis del pigmento pardo, la melanina, que después es transferido a los queratinocitos adyacentes. El melanoma maligno es un tumor cuya incidencia está aumentando de forma dramática entre las personas de piel blanca de todo el mundo. Los melanomas malignos se diseminan inicialmente desde los vasos linfáticos a los ganglios linfáticos regionales y posteriormente por vía sanguínea, por lo que el control a tiempo de la enfermedad es extremadamente difícil.
Datos epidemiológicos y moleculares sugieren la existencia de una estrecha asociación entre el desarrollo de tumores de piel no-melanoma y una excesiva exposición a la radiación ultravioleta de la luz solar9,10. Sin embargo esta asociación tan directa no está totalmente clara con respecto al origen del melanoma, en el cual múltiples factores parecen intervenir: predisposición genética, exposición a la luz ultravioleta (sol, fuentes artificiales), y exposición ambiental a mutágenos (sustancias químicas, virus, radiaciones), entre otros11.
Como consecuencia de una exposición repetida y continuada a la radiación ultravioleta ocurren modificaciones en el DNA que de forma natural son reparadas por los mecanismos celulares de escisión de nucleótidos (NER, nucleotide-excision repair) y de escisión de bases (BER, base-excision repair)12. El efecto acumulativo de modificaciones no reparadas puede desencadenar la aparición de mutaciones puntuales en genes diana como el gen supresor de tumores p53, el cual no está únicamente alterado en cánceres de piel sino en tumores de diferente histología13. Constituyendo p53 una diana clave para el inicio del proceso neoplásico de piel.
Características de la radiación ultravioleta (UV)
La luz solar es energía radiante electromagnética compuesta principalmente por el espectro de luz ultravioleta (100 a 400 nm), luz visible (400 a 760 nm) e infrarroja (760 a 1.800 nm), aunque también están presentes longitudes de onda corta (ionizantes), y onda larga (microondas y radiofrecuencia). Estas radiaciones son modificadas de manera importante por su paso a través de la atmósfera y solamente dos tercios de esta energía penetra en la Tierra.
La radiación UV se divide en tres bandas: UVA (320 a 400 nm), UVB (280 a 320), y la UVC (200 a 280 nm). La UVA no es filtrada por la capa de ozono en el mismo grado que la UVB y la UVC, y cantidades suficientes de la misma penetran a través de las nubes y de los vidrios14. En un día de verano, la UVB comprende aproximadamente el 5% de la radiación UV, y la UVA el 95% restante. Sin embargo la UVB es más responsable que la UVA en producir daño biológico, ya que contribuye con cerca del 80% de los efectos dañinos que se asocian a la exposición solar, la UVA sólo produce el 20% restante15. La cantidad de radiación UV y luz visible que alcanzan un cierto nivel de la piel varía con su longitud de onda. En general, las longitudes de onda largas penetran más profundamente, lo cual se puede explicar por las propiedades ópticas de la piel. Cuando la luz visible y la UV alcanzan la piel, parte es reflejada, parte es absorbida, y parte es transmitida a diferentes capas de células, hasta que la energía del rayo incidente se disipa. La porción de luz que es absorbida por las moléculas en los tejidos es la más importante ya que se trata de la energía que puede causar respuestas tisulares16.
La radiación es absorbida por moléculas en la piel denominadas cromóforos, los cuales pueden ser endógenos (por ejemplo, el DNA, la melanina, el ácido urocánico, pequeños péptidos, y el colesterol) o exógenos (drogas fotosensibilizantes) capaces de inducir una respuesta fotobiológica, como una quemadura solar o una fotosensibilidad inducida por drogas16. Los niveles de penetración de los rayos UVB y UVA a nivel de la piel son diferentes. El 70% de la radiación UVB es absorbida por el estrato córneo de la epidermis, a diferencia de la radiación UVA que es absorbida entre el 70-80% por células de la dermis y melanocitos de la epidermis basal.
Modelo de progresión tumoral en cáncer de piel
Durante la formación de un cáncer de piel se atraviesan tres estadios: inicio, promoción y progresión, en los cuales está implicada la radiación UV como agente carcinogénico. Durante la fase inicial, los fotoproductos no-reparados originados por efecto de la radiación UV pueden ocasionar mutaciones en regiones codificantes de oncogenes y genes supresores de tumor. La exposición UV crónica da lugar a la aparición de un tumor benigno (como la queratosis actínica)17 formado a partir de la expansión clonal de células epidérmicas portadoras de modificaciones en diferentes genes como el proto-oncogen ras o el gen supresor de tumor p53. Una irradiación UV continuada permite la progresión tumoral mediante la selección de clones de células resistentes a la apoptosis. El papel central que tiene la inactivación de p53 en la carcinogénesis de piel fue por primera vez demostrado por los hallazgos de Jiang y colaboradores18, encontrando que aquellos ratones que eran defectivos para p53, p53-/- o p53+/- bajo inducción con radiación UV desarrollaban más tempranamente tumores de piel que los ratones salvajes. La exposición solar prolongada tiene además un segundo efecto carcinogénico, y es la pérdida de la interacción Fas-L/Fas como consecuencia de la acumulación de mutaciones en p5319.
Efectos de la radiación UV. Daño producido en el DNA
Los sistemas de reparación del DNA juegan un papel crucial en el mantenimiento de la integridad del genoma contra agentes genotóxicos que son los responsables del desarrollo tumoral20. Esta asociación se pone de manifiesto por la alta incidencia de tumores de piel que presentan los pacientes afectados por síndromes con defectos en el sistema de reparación de escisión de nucleótidos (NER), como el caso del xeroderma pigmentosum (XP), el síndrome de Cockayne (CS), y la tricotiodistrofia (TTD)21. En estos síndromes, numerosas lesiones producidas por la radiación UV no reparadas dan lugar a la aparición de mutaciones en genes clave que posteriormente desencadenan tumores de piel (melanoma y no-melanoma).
Los daños generados en el DNA son reparados por diversos mecanismos (Fig. 1). Así, las roturas de la doble hélice son reparadas por un mecanismo de reparación dependiente de recombinación homóloga, y muchas de las pequeñas modificaciones de bases son eliminadas por el sistema de reparación de escisión de bases (BER)12. El sistema de reparación por escisión de nucleótidos (NER) elimina distorsiones voluminosas en la doble cadena de DNA22. Existiendo una considerable especificidad de sustrato de las vías reparadoras, con proteínas que son comunes a las diferentes vías23. La radiación ultravioleta induce la formación de dos fotoproductos relevantes, los dímeros de pirimidina tipo cilobutano (CPDs), y los fotoproductos de 6-4 pirimidina pirimidona (6-4 PPs)24, 25. Ambas lesiones se forman exclusivamente en dímeros de pirimidinas, constituyendo los "hot spots" o puntos calientes de mutación inducidos por radiación UV26, 27. Ambas lesiones son reparadas por el sistema NER, aunque los productos 6-4 PPs se reparan unas 5 veces más rápido que los CPDs, sin embargo las lesiones CPDs son mucho más abundantes en el genoma. Esta diferencia puede deberse a que los fotoproductos 6-4 PPs producen una distorsión mayor en la doble hélice de DNA y por ello son más evidentes para los sistemas de reparación, a diferencia de los fotoproductos CPDs. El sistema de reparación NER implica la acción de unas 20 a 30 proteínas que actúan de forma secuencial durante el reconocimiento del daño. Producen apertura local de la doble hélice en el punto de la lesión, e incisión de la hebra dañada en uno de los lados de la lesión. Tras la escisión del oligonucleótido que contiene el daño, la mella resultante se rellena mediante la actividad de la DNA polimerasa correspondiente.
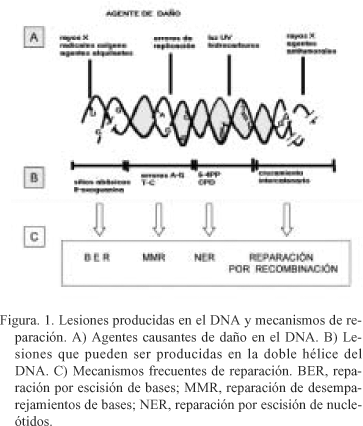
Estas lesiones únicas igualmente dan lugar a mutaciones únicas en el DNA. La radiación ultravioleta induce predominantemente transiciones C®T y CC®TT en las secuencias de dímeros de pirimidina, constituyendo la característica más destacada de la mutagénesis inducida por la radiación UV28. Ambas mutaciones se piensa que se originan durante la replicación semiconservativa del DNA, cuando la DNA polimerasa llega a una lesión de dímero de pirimidina no sabe interpretar que base complementaria debe de insertar, con lo cual la enzima por defecto introduce un adenina29. Por lo tanto los dímeros C-C pueden originar mutaciones del tipo C®T o bien CC®TT, ya que la DNA polimerasa inserta una A enfrente de una C de la hebra complementaria. Posteriormente durante la replicación semiconservativa del DNA se insertan residuos de T en la hebra recién sintetizada dando lugar a la aparición de la mutación.
Incremento en los niveles de la proteína p53
El gen supresor de tumores p53 codifica una fosfo-proteína de 53-kDa que ayuda a mantener a las células en su estado de no-malignidad a través del control que ejerce sobre el ciclo celular mediante la activación transcripcional de genes reguladores30. Una gran variedad de agentes que producen daño en el DNA inducen elevados niveles de p53 (Fig. 2). Lo que conduce a la detención del ciclo celular, permitiendo a los mecanismos de reparación celulares eliminar las lesiones en el DNA antes de que ocurra la síntesis de DNA durante la fase S del ciclo celular31, o bien se induce apoptosis en aquellas células que presentan excesivo daño en el DNA32. Además de estas funciones p53 puede directa o indirectamente modular la reparación del DNA33 (Fig. 2).
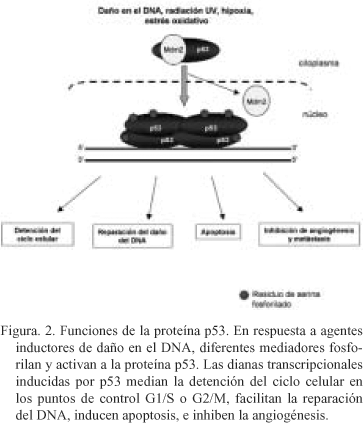
Estudios realizados tanto en líneas celulares, epidermis de ratón, y tejido humano han mostrado que los daños ocasionados en el DNA por efecto de la radiación UV inducen la expresión de la proteína p5319, 34-35. La forma en la cual las roturas de la doble hélice de DNA inducen la expresión de p53 no es del todo conocida, aunque parecen estar implicadas cascadas de quinasas asociadas a otras proteínas. De hecho, la radiación UV induce la fosforilación y activación de la proteína p53 en múltiples residuos de serina, incluyendo Ser 15, 20, 33, 37, 46, y 39236. Igualmente existe evidencia de la actuación de diferentes proteínas durante el proceso de fosforilación de p53, incluyendo: ATM, ATR, p38, y las quinasas de mitógeno activadas (MAP-quinasas)37-40. La fosforilación de p53 y/o de su regulador negativo Mdm2 activa a p53 a través de diferentes mecanismos: a) estabilizando a p53 mediante la ruptura de la interacción con Mdm2; b) aumentando su actividad transcripcional; y c) promoviendo la localización nuclear de p53.
Inducción de mutaciones de p53 en tumores de piel
La aparición de mutaciones en p53 parece ser un evento temprano en el desarrollo de un cáncer de piel inducido por la radiación UV. De hecho, en la piel normal expuesta a la luz solar aparecen miles de clones celulares mutados para p5341. Igualmente las lesiones premalignas como la queratosis actínica presentan una elevada frecuencia de mutaciones en p5342. En estudios recientes se ha puesto de manifiesto además que las áreas de piel adyacentes a un tumor de piel exhiben mutaciones de p53 con características distintas de las encontradas en la lesión tumoral43. Esto podría sugerir que sólo un subtipo de mutaciones de p53 inducidas por la radiación UV confiere malignidad a las células43. En estudios realizados con modelos murinos de cáncer de piel inducida con radiación UV se ha encontrado que las mutaciones de p53 ocurren muy tempranamente durante el desarrollo tumoral44. En estos modelos las transiciones C®T representan alrededor del 70% de las mutaciones encontradas en p53, mientras que las transiciones CC®TT representan solamente el 10-20%44.
En los tumores de piel humanos, las mutaciones consistentes en transiciones C®T son las más frecuentes y aparecen concretamente en secuencias de tri-nucleótidos 5'-PyCG-3' (Py, pirimidina) del gen p53. Se trata de secuencias génicas que en el DNA de los queratinocitos de piel se encuentran metiladas (5-metil-citosina, 5-m-C). De los diferentes "hot spots" que aparecen mutados en la proteína p53, siete de ellos contienen citosinas metiladas en las secuencias 5'-CCG-3' (Codones: 152, 158,196, 213, 245, 248, y 282)45. Únicamente se ha encontrado un punto "hot spot" común a los tumores no-melanoma y melanoma, la Arginina 248. Este residuo de arginina es codificado por una secuencia -CCG- metilada, siendo un aminoácido que se encuentra en la superficie de la proteína p53 e interacciona directamente con el DNA, por tanto altera las funciones de transactivación de p53.
5-metil-citosina y mutagénesis producida por la radiación UV
Los dímeros de pirimidina tipo ciclobutano (CPDs) que se forman preferentemente bajo inducción con la radiación UV, se originan principalmente en las secuencias 5'-PyCG-3', con la citosina en 5' metilada. El aumento de la aparición de mutaciones en secuencias del tipo CpG metiladas es atribuible a dos mecanismos diferentes (Fig. 3). Una ruta implica la actuación de la enzima DNA polimerasa propensa a error que incorpora adeninas enfrente de citosinas o 5-metil-citosinas dentro del dímero de pirimidina tipo ciclobutano (Fig. 3). En la segunda ruta, la citosina o 5-metil-citosina del dímero de dipirimidina es primero desaminada convirtiéndose en timina en una primera reacción de desaminación y en uracilo en una segunda desaminación, posteriormente intervendría la enzima DNA polimerasa h libre de error que introduce la base complementaria (Fig. 3).
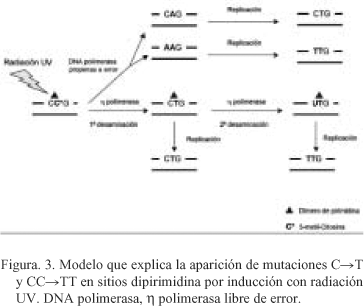
Puntos "hot spots" de mutación de p53 encontrados en los carcinomas basocelulares, espinocelulares, y melanoma maligno
Los codones que suelen aparecer mutados en los tumores basocelulares son el 177, 196, y 24545. El codón 177 es bastante específico de los tumores basocelulares y no suele encontrarse mutado en otros tipos tumorales. Parece ser que este codón se repara muy lentamente bajo la inducción con radiación UV46. El codón 196 se ha encontrado igualmente mutado en tumores de mama y colon, y el codón 245 en tumores de pulmón, cabeza y cuello, ovario, y estómago46. En los tumores espinocelulares la mutación en el codón 278 parece ser muy específica. Aunque igualmente aparece este codón mutado en otros tumores sólidos, sin embargo con una frecuencia muy baja46. En el melanoma maligno aparecen mutados los codones 104 (raramente mutado en otros tipos de tumores), 213, 286, 290, y 29645.
Los tumores no-melanoma difieren notablemente del melanoma maligno, no sólo con respecto a su origen sino también en el tipo de mutaciones que aparecen en p53. En el melanoma maligno únicamente aparece un 10% de mutaciones en el gen p5311, a diferencia de los tumores basocelulares y espinocelulares con más del 60% de mutaciones en p5345. Estos datos sugieren que existe una vía independiente de p53 implicada en el desarrollo tumoral del melanoma.
Estudios estadísticos que analizan el espectro de mutaciones de p53 en tumores tipo melanoma y no-melanoma revelan la existencia de diferencias significativas (p<0.05) entre ambos grupos tumorales. El melanoma maligno se caracteriza por una elevada frecuencia de transiciones A®G47, probablemente originadas por otros agentes mutagénicos diferentes de la radiación UV. Este tipo de sustituciones pueden ser debidas a la formación de radicales libres del oxígeno. Sin embargo no existen diferencias estadísticas entre el tipo de mutaciones encontradas en los tumores basocelulares y espinocelulares que exhiben la huella característica de mutaciones inducidas por la radiación UV en sitios dipirimidina.
Conclusión
En los últimos años se ha producido un incremento notable de la incidencia de cáncer de piel. Existiendo una relación directa entre ese aumento y la exposición reiterada al componente ultravioleta de la luz solar. Si bien en los tumores no-melanoma esa asociación parece ser directa, no es tan aparente en el melanoma maligno. La huella característica que presenta la mutagénesis inducida por la radiación UV en los tumores no-melanoma es la formación de dímeros de pirimidina. Los cuales mayoritariamente conducen a la aparición de transiciones C®T y CC®TT en genes diana claves. Entre estos genes p53, "el guardián del genoma", es la primera diana implicada en el desarrollo tumoral. La acumulación de mutaciones inducidas por la radiación UV en p53 no reparadas, hace que las células sean resistentes a los mecanismos de apoptosis y puedan llegar a malignizarse.
Bibliografía
1. Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol 1994; 30:774-778. [ Links ]
2. Houghton AN, Polsky D. Focus on melanoma. Cancer Cell 2002; 2:275-278. [ Links ]
3. Goldberg LH. Basal cell carcinoma. Lancet 1996; 347:663-667. [ Links ]
4. Black HS, deGruijl FR, Forbes PD, et al. Photocarcinogenesis: an overwiew. J Photochem Photobiol B 1997; 40:29-47. [ Links ]
5. Wong CS, Strange RC, Lear JT. Basal cell carcinoma. BMJ 2003; 327:794-798. [ Links ]
6. Kane CL, Keehn CA, Smithberger E, Glass LF. Histopathology of cutaneous squamous cell carcinoma and its variants. Sem Cutan Med Surg 2004; 23:54-61. [ Links ]
7. Struijk L, Ter Schegget J, Bouwes-Bavinck JN, Feltkamp MC. Human papillomavirus in the aetiology of skin cancer. Ned Tijdschr Geneeskd 2005; 149:518-522. [ Links ]
8. Anwar J, Wrone DA, Kimyai-Asadi A, Alam M. The development of actinic keratosis into invasive squamous cell carcinoma: evidence and evolving classification schemes. Clin Dermatol 2004; 23:189-196. [ Links ]
9. Ananthaswamy HN, Pierceall WE. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochem Photobiol 1990; 52:1119-1136. [ Links ]
10. de Gruijl FR. Skin cancer and solar UV radiation. Eur J Cancer 1999; 35:2003-2009. [ Links ]
11. Zerp SF, van Elsas A, Peltenburg LTC, Schrier PI. p53 mutations in human cutaneous melanoma correlate with sun exposure but are not always involved in melanogenesis. Br J Cancer 1999; 79:921-926. [ Links ]
12. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366-374. [ Links ]
13. Hainaut P, Soussi T, Shomer B, et al. Database of p53 gene somatic mutations in human tumors and cell lines: updated compilation and future prospects. Nucleic Acids Res 1997; 25:151-157. [ Links ]
14. Wharton J, Cockerell CJ. The sun: a friend and enemy. Clin Dermatol 1998; 16:415-419. [ Links ]
15. Diffey BL. Ultraviolet radiation and human health. Clin Dermatol 1998; 1:83-89. [ Links ]
16. Kochevar I. Photobiology. Basic science. Dermatol Clin 1986; 4:171-179. [ Links ]
17. Ziegler A, Jonason AS, Leffel DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature 1994; 372:773-776. [ Links ]
18. Jiang W, Ananthaswamy HN, Muller HK, Kripke ML. p53 protects against skin cancer induction by UV-B radiation. Oncogene 1999; 18:4247-4253. [ Links ]
19. Ouhtit A, Gorny A, Muller HK, Hill LL, Owen-Schaub LB, Ananthaswamy HN. Loss of Fas-ligand expression in mouse keratinocytes during UV carcinogenesis. Am J Pathol 2000; 157:1975-1981. [ Links ]
20. Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer 2001; 1:22-33. [ Links ]
21. Lehmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003; 85:1101-1111. [ Links ]
22. de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev 1999; 13:768-785. [ Links ]
23. Lindahl T, Wood RD. Quality control by DNA repair. Science 1999; 286:1897-1905. [ Links ]
24. Mitchell DL. The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem Photobiol 1988; 48:51-57. [ Links ]
25. Mitchell DL, Nairn RS. The biology of the 6-4 photoproduct. Photochem Photobiol 1989; 4:805-819. [ Links ]
26. Sage E. Distribution and repair of photolesions in DNA: genetic consequences and the role of sequence context. Photochem Photobiol 1993; 57:163-174. [ Links ]
27. Mitchell DL, Jen J, Cleaver JE. Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acid Res 1992; 20:225-229. [ Links ]
28. Pfeifer GP, You YH, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res 2005; 571:19-31. [ Links ]
29. Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA 1991; 88:10124-10128. [ Links ]
30. Harris CC. Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J Natl Cancer Inst 1996; 88:1442-1455. [ Links ]
31. Zhan Q, Carrier F, Fornace AJ Jr. Induction of cellular p53 activity by DNA damaging agents and growth arrest. Mol Cell Biol 1993; 13:4242-4250. [ Links ]
32. Yonish-Rouach E, Reznitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild type p53 induces apoptosis of myeloid leukemic cells that is inhibited by IL-6. Nature 1991; 352:345-347. [ Links ]
33. Li G, Mitchell DL, Ho VC, Tron VA. Differentiation-dependent p53 regulation of nucleotide excision repair in keratinocytes. Am J Pathol 1997; 150:1457-1464. [ Links ]
34. Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in non-transformated mouse cells. Mol Cell Biol 1984; 4:1689-1694. [ Links ]
35. Ouhtit A, Muller HK, Davis DW, Ullrich SE, McConkey D, Ananthaswamy HN. Temporal events in skin injury and the early adpatative responses in ultraviolet-irradiated mouse skin. Am J Pathol 2000; 156:201-207. [ Links ]
36. Kapoor M, Hamm R, Yan W, Taya Y, Lozano G. Cooperative phosphorylation at multiple sites is requiered to activate p53 in response to UV radiation. Oncogene 2000; 19:358-364. [ Links ]
37. Bannin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998; 281:1674-1679. [ Links ]
38. Tibbetts RS, Rrumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 1999; 13:152-157. [ Links ]
39. Milne DM, Campbell LE, Capmbell DG, Meek DW. P53 is phosphorylated in vitro and in vivo by an ultraviolet radiation-induced protein kinase characteristic of the c-Jun kinase, JNK1. J Biol Chem 1995; 270:5511-5518. [ Links ]
40. She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylated p53 protein at serine 15 in response to UV radiation. J Biol Chem 2000; 275:20444-20449. [ Links ]
41. Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA 1996; 93:14025-14029. [ Links ]
42. Ziegler A, Jonason AS, Leffel DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature 1994; 372:773-776. [ Links ]
43. Ren ZP, Hendrum A, Potten F, et al. Human epidermal cancer and accompanying precursors have identical p53 mutations different from p53 mutations in adjacent areas of clonally expanded non-neoplastic keratinocytes. Oncogene 1996; 12:765-773. [ Links ]
44. Ananthaswamy HN, Loughlin SM, Cox P, Evans RL, Ullrich SE, Kripke ML. Sunlight and skin cancer: inhibition of p53 mutations in UV-irradiated mouse skin by sunscreens. Nat Med 1997; 3:510-514. [ Links ]
45. Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Human Mutation 2003; 21:217-228. [ Links ]
46. Tornaletti S, Pfeifer GP. Complete and tissue-independent methylation of CpG sites in the p53 gene: implications for mutations in human cancers. Oncogene 1995; 10:1493-1499. [ Links ]
47. Soto JL, Cabrera CM, Serrano S, López-Nevot MA. Mutation analysis of genes that control the G1/S cell cycle in melanoma: TP53, CDKN1A, CDKN2A, and CDKN2B. BMC Cancer 2005; 5(1):36. [ Links ]
 Correspondencia:
Correspondencia:
Dra. C. M. Cabrera
Servicio de Anatomía Patológica
Hospital Universitario Virgen de las Nieves
Edificio de Gobierno 4ª Planta
Avenida Fuerzas Armadas, 2
E-18014 Granada
mcabrm@fundacionhvn.org
Recibido: 11.11.05
Revisado: 22.12.05
Aceptado: 13.01.06

