










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 n.1 Madrid Jan. 2004
| POINT OF VIEW |
Molecular genetics of colorectal cancer
D. Cruz-Bustillo Clarens
National Institute of Gastroenterology. Havana, Cuba
ABSTRACT
Colorectal tumours constitute an excellent system to study carcinogenesis and the molecular events implicated in the development of cancer. Attending to the way it is transmitted, colorectal cancer may appear in one of three forms: sporadic, familial, and hereditary. The sporadic form is most common and has no familial or hereditary associated factor thus far, while familial and hereditary forms show the same inheritance pattern. Hereditary colorectal cancers develop by means of defined stages that go from lesions in the crypt of the colon through adenomas to manifest cancer. They are characterised by the accumulation of multiple mutations in tumour suppressor genes and oncogenes that affect the balance between cell proliferation and apoptosis. The colorectal carcinogenesis pathway is not unique and there are probably several ways for the initiation, development and progression of colorectal tumours.
Key words: Colorectal cancer. Colorectal carcinogenesis. Tumour suppressor genes. Caretakers and gatekeepers. Germinal mutation. Sporadic mutation. Microsatellite instability.
Cruz-Bustillo Clarens D. Molecular genetics of colorectal cancer. Rev Esp Enferm Dig 2004; 96: 48-59.
Recibido: 21-02-03.
Aceptado: 22-07-03.
Correspondencia: Diana Cruz-Bustillo Clarens. Calle 25 #503 entre H e I, Vedado. 10400 La Habana, Cuba. Phone: (537) 832 5067. Fax: (537) 333 253. e-mail: dcruzb@infomed.sld.cu
INTRODUCTION
In recent years extraordinary advances have been accomplished in colorectal carcinogenesis research.
Colorectal tumours constitute an excellent system to study both carcinogenesis and the molecular events involved in the development of cancer. Due to the increasing clinical and histopathological information in relation to malignant colorectal tumours, it is currently a fact that the majority of, if not all, colorectal cancers (CRC) come from previous benign tumours, namely adenomas. Another advantage of the study of carcinogenesis using CRC as a model is that it is possible to obtain study materials from a range of very small adenomas to fully developed tumours with metastasis. On the other hand, both hereditary and environmental factors contribute to the development of CRC, all of which enable the investigation of somatic and hereditary genetic alterations.
CLASSIFICATION OF COLORECTAL CANCERS
Attending to the way it is transmitted, there are three major forms of colorectal cancer:
1. Sporadic.
2. Familial.
3. Hereditary.
The proportion of each one in the population varies (Fig. 1). Hereditary forms and hamartomatous syndromes are least common (1). Ten percent to 30% of cases correspond to familial risk, and the rest to sporadic colorectal cancer.
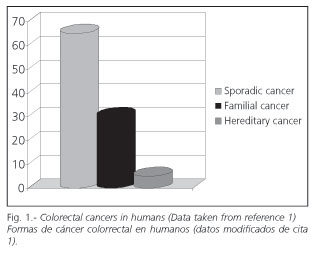
The term sporadic is usually used to differentiate cancers occurring in individuals who do not carry a mutation conferring tumour susceptibility from cancers occurring in individuals who carry a known mutation associated with this disease. This difference is not absolute because the genetic factor seems to influence the probability of cancer onset even in the presence of a specific mutation. The term sporadic is also used to describe cancer in individuals lacking a family history of cancer (2). The vast majority of cancers are considered sporadic.
So far, no gene associated to familial cancers has been identified. However, population studies show a two to three-fold greater risk of CRC versus the normal population when first degree relatives have sporadic cancer in the colon. Results of multiple family studies indicate that family risk is the probable outcome of an inherited susceptibility with partial penetrance to develop adenomas and colon cancer (3).
Inherited factors may determine the susceptibility of an individual to develop adenomas and colon cancer, while environmental factors most probably determine which of the genetically predisposed individuals will develop small adenomas, big adenomas, and finally colorectal cancer (4).
The most common hereditary forms of CRC are familial adenomatous polyposis (FAP) and hereditary non-polyposis colorectal cancer (HNPCC), although there are other syndromes associated with predisposition.
HEREDITARY SYNDROMES PREDISPOSING TO COLORECTAL CANCER
In 1998, Lynch and Lynch published a most complete classification of hereditary disorders predisposing to CRC (5).
These authors not only classified the different cancer syndromes, but also described their pattern of inheritance and associated germinal mutations; polyp information; other associated cancers; non-cancerous characteristics of the syndrome; population screening; surgical and/or prophylactic procedures; DNA testing in pre-symptomatic individuals, and genetic counselling.
According to these authors the individual syndromes predisposing to CRC are:
Familial adenomatous polyposis (FAP).
Attenuated familial adenomatous polyposis (AFAP).
I1307K mutation in Ashkenazi Jews.
Juvenile polyposis coli.
Peutz-Jeghers syndrome.
Slight adenomatous polyposis of the colon and Burts colorectal cancer.
Hereditary non-polyposis colorectal cancer (HNPCC).
Familial colorectal cancer
Familial ulcerative colitis and Crohns disease.
With the exception of the latter two, the rest are transmitted in a dominant autosomal way.
FAP has a very characteristic clinical phenotype, represented by profuse polyposis. It has been described (6) as an autosomal dominant disorder typically presented with colorectal cancer at early ages, secondary to extense adenomatous polyposis of the colon. Polyps may also appear in the upper gastrointestinal tract, and tumours may occur in other locations, including the brain and thyroid. Other clinical characteristics include: retinal lesions named congenital hypertrophy of the retinal pigment epithelium (CHRPE), jaw cysts, sebaceous cysts, and osteomas. FAP manifests in patients who inherit germinal mutations in the APC gene, which is localized in 5q21.
There are several classifications of FAP, one of them by number of polyps (7). The disease is classified as mild when there are fewer than a thousand polyps in the intestine, while it will be severe if this amount is exceeded.
Gardners syndrome is a phenotypic variant of FAP that also includes extracolonic features such as epidermoid cysts, jaw osteomas, CHRPE, fibromas, and desmoid tumours (8-10).
The average onset age for FAP is 39 years although the disease is sometimes expressed during puberty and around 20 years of life. Other extracolonic cancers associated with FAP are: stomach, small intestine, and periampullar carcinoma and sarcoma (5).
AFAP differs in that adenomas may be flat and predominate in the proximal colon; their major characteristic is that they are scarce (5 to 10), but sometimes over 100. Cancer develops at older ages (50 years average), and polyps in the fundic glands of the stomach and adenomas of the duodenum may be associated. (5).
Turcots syndrome is characterized by profuse adenomas in the colon (from 50 to over 100) and by the development of cancers in the central nervous system (CNS), specifically the brain. There are two variants depending on the mutated gene. The first variant presents germinal mutations in the APC gene, and in this disease medulloblastomas predominate; the second one develops mutations in the hMLH1 and hPMS2 genes, and multiform glioblastoma predominates (5).
Juvenile polyposis is characterized by the difuse presence of 10 or more juvenile hamartomatous polyps in the colon, which may develop also in the stomach and small intestine. It is molecularly identified by mutations in the gene coding for tyrosine-phosphate protein (PTEN) (1,5).
Peutz-Jeghers syndrome is characterized by the presence of hamartomatous polyps in the stomach, small intestine, and colon. It also develops muco-cutaneous pigmentation of melanin usually in the perioral region, and is accompanied by mutations in the gene coding for serine-threonine kinase (STK11) on chromosome 19p13.3 (1).
HNPCC presents adenomas only occasionally, never profusely. Its diagnosis requires the exclusion of FAP. Adenomas are generally bigger and more vellous; their frequency of appearance is the same as in the normal population. It is characterized by the development of CRC at early ages, predominant in the right colon, with an excess of synchronic and metachronic tumours. Other cancers associated with HNPCC are: endometrium, ovary, small intestine, stomach, ureter and renal pelvis. The average age at onset is 44 years, and a rapid progression adenoma-carcinoma is observed. The Muir-Torre syndrome is a rare variant presenting CRC, sebaceous adenomas, epitheliomas, and other skin lesions (5).
In 1991, the Amsterdam Criteria were established (11) to confirm the clinical diagnosis of HNPCC families. Since these criteria are rather restricted, several modifications have been proposed and currently the Bethesda Criteria, established in 1997 (12) (Tables I and II) and the Amsterdam Criteria II, which extend the tumour spectrum including cancers in endometrium, small intestine, renal pelvis and ureter, are also available (13).


HNPCC is molecularly characterized by germinal mutations in the genes of the DNA mismatch repair system, mainly hMLH1 and hMSH2, and also because the colorectal tumours show microsatellite instability (5).
THE ADENOMA-CARCINOMA SEQUENCE
The cells of the normal intestine mucosa have a polyclonal origin and they develop from a variety of stem cells, while CRC cells are monoclonal (14).
CRC develops through definite stages that go from lesions in the colonic crypts through adenomas to cancer. This adenoma-carcinoma sequence is characterized by the accumulation of multiple mutations in tumour suppressor genes and oncogenes that affect the balance be-tween cell proliferation and apoptosis (Fig. 2). A modern explanation for tumour genesis is based on the concept that each of these mutational events confers a growth advantage to each tumour cell (14,15). And every following event confers the cell additional growth advantages compared to the rest of tumour cells, resulting in multiple staged clonal expansion and, eventually, tumour progression (14,16).

Mutational events include small deletions, insertions or substitutions of only one nucleotide, but major genetic changes may occur, like gene amplification or complete chromosome loss. These alterations may affect two different classes of genes: proto-oncogenes and tumour suppressor genes. The majority of hereditary forms of CRC present alterations in tumour suppressor genes (14).
A more updated classification (17) subdivides tumour suppressor genes into two classes: gatekeepers and caretakers. Gatekeepers directly inhibit tumour growth and promote apoptosis, while inactive caretakers do not affect directly tumour growth, but they result in genomic instability thus contributing to the general increase of mutation rate and acceleration of cancer development.
THE APC GENE
The adenomatous polyposis coli (APC) gene is the authentic gatekeeper of cell proliferation in the colon epithelium.
The APC gene was identified and characterized in 1991 (18, 19). It is located in chromosome 5q21 and is made up of 8535 bp distributed into 15 exons, although in 1996 Thliveris (20) described it as a gene divided into 21 exons. A recent description includes a 10A exon. This gene codes for a large protein of 2843 aminoacids in its more common isoform (21). Exon 15 occupies >75% of the coding sequence of APC, and is the most common mutational region, both for germinal and somatic mutations (22).
The APC protein forms homo-oligomers, associates itself with catenins, and expresses itself in various types of tissue. It is made up of an oligomerization domain and an armadillo region in the amino-terminal end, 15 or 20 aminoacid repetitions in its central portion, and a carboxi-terminal end containing a basic domain and union sites for other proteins. The multiple domains of APC allow interaction with other proteins. Each domain has a specific function regarding the proteins activity (23).
The oligomerization domain allows the protein to form homodimers, the proteins active form (24), while the armadillo region seems to allow the protein to play a role in the stabilization and motility of the cytoskeleton, even though it must not be essential for APCs role as a tumour suppressor gene (23). The 15-aminoacid repetitions confer the protein union sites for ß-catenin, and the 20-aminoacid repetitions are the union site for this protein. This site has been found essential for the binding of ß-catenin (25).
The more frequent mutations in the APC gene produce inactive truncated proteins. The basic domain permits APC to bind to microtubules (23)
The APC protein has various functions (4):
1. Regulation of ß-catenin-induced signalling.
2. Regulation of cell adhesion via ß-catenin and E-cadherin.
3. Regulation of cell migration via interaction with microtubules.
4. Cell cycle block perhaps by direct inhibition of cell cycle components.
5. Coordinated regulation of cell adhesion and motility (26).
Wnt SIGNALLING PATHWAY AND CELL PROLIFERATION
The development of embryonic tissues and organs is controlled by several signalling pathways that interact with each other to offer information and to induce the specification of cellular fate. One of the main signalling systems is the Wnt pathway (25). Wnt (wingless) proteins direct the differentiation of various types of cells in insect and vertebrate embryos.
These proteins constitute a family of highly preserved signalling molecules that regulate intercellular interactions during embryogenesis. Wnt genes have also been proved to be involved in cancer (28).
It has been demonstrated that Wnt signalling is essential to maintain the stem-cell compartment within intestinal crypts (29). Stem cells are mutipotent cells found in many tissues, and they can meet a variety of fates. When they are exposed to selected growth factors and cytokines, they generate progenitor cells which transiently proliferate and then leave the cell cycle to ultimately differentiate (30).
Wnt signalling proteins participate in various tissues, as the skin, adipose tissue, haematopoietic tissue, and others (25).
THE ROLE OF APC AND ß-CATENIN IN THE Wnt SIGNALLING PATHWAY
The APC protein plays a substantial role in the regulation of the Wnt signalling pathway. Usually, APC will form a complex with ß-catenin, axin, glycogen synthase kinase 3ß (GSK 3ß) and other proteins. Axin contains binding domains for the essential components involved in ß-catenin degradation, and GSK 3ß phosphorylates ß-catenin in its serine and threonine residues (31).
The marking of ß-catenin is sufficient for its immediate ubiquitin-dependent proteolysis (32). When the complex is not formed, either due to a Wnt signal, the inactivation of APC, or a mutation of ß-catenin, the latter is neither phosphorylated nor degraded, but it is accumulated in the cytoplasm and nucleus (33,34). There, it associates with members of the T cellular factor (TCF) and the gene transcription activator family implicated in cell proliferation, for example, oncogenes c-myc and D1 cyclin. Both proteins regulate the cell cycle (35-38).
ROLE OF APC IN INTERCELLULAR ADHESION
The fact that APC binds to ß-catenin implies that APC also plays a role in epithelial cellular adhesion (39). Catenins associate with cadherins, which are involved in intercellular adhesion (39). This binding is a critical structure of the adherens zone complex that participates in adhesion and in intercellular communication. E-cadherin serves also as an anchorage area to the actin cytoskeleton (16). E-cadherin is responsible for cell-cell adhesion in epithelial cells and thus, it is essential that catenin binds one of its cytoplasmatic domains (40). This domain has an identical motif to ß-catenins in APC. E-cadherin and ß-catenin interaction is regulated by the phosphorylation of the latter, and this phosphorylation only takes place when a complex with the homodimer of APC protein has previously been formed (23). On the other hand, APC contributes to the ordered migration of intestinal cells inside the crypt, and ß-catenin plays a crucial role in this function (41).
EVENTS IN THE COLONIC CRYPT
The microarchitecture of the colon is characterised by crypts that are approximately 50 cells deep. In the small bowel, crypts and villi provide a very large surface area for nutrient absorption. In the large bowel this great area is not necessary because absorption is mainly restricted to water (4).
In the normal colon epithelium, there is an almost constant and normal renewal of the superficial epithelium, approximately every six days, through cell proliferation and differentiation in the crypt. Colonocyte proliferation takes place in the lower portion of the crypt and is characterized by mitosis and because colon cells migrate to the upper part of the crypt moving away from stem cells. Differentiation and maturation of newly born cells take place while these migrate along the crypt (4). Mature cells lose their capacity to divide again, and finally die by apoptosis and exfoliate to the lumen.
In the adenoma, this sequence is altered. Continued mitosis takes place and cells do not differentiate, so the compartment where they proliferate may take up the whole crypt. An adenoma is a benign neoplasm, and even though it is generally accepted that colon cancers rise from colon adenomas, it is a well known fact that the majority of adenomas do not develop into carcinomas. The exact frequency of this progression from an adenoma to cancer is unknown yet (16).
MOLECULAR MODEL OF COLORECTAL CARCINOGENESIS
Survivin is a member of the antiapoptotic family of proteins (42) which appears to be overexpressed in cancers but not in the corresponding normal adult tissues (43). Apoptosis is the normal mode of programmed cell death. On the other hand, it has been suggested that survivin could participate in cell division regulation (44), specifically in mitosis, and that it might play a dominant role in microtubule function (45).
Let us take a look at the molecular events taking place in the colonic crypt and hypothesize how colorectal carcinogenesis takes place.
Survivin expresses itself preferentially in the lower part of the normal colonic crypt, where stem cells responsible for cellular proliferation reside. Thus, in the base of the crypt the proliferation of stem cells which migrate up the crypt wall while they differentiate and mature takes place, until they reach the apex of the crypt. Therefore, the function of survivin in the base of the crypt is to inhibit apoptosis so that all the necessary cells, substituting those that will be eliminated in the apex and ultimately exfoliate to the lumen, may regenerate. On the contrary, survivin will be responsible for conferring stem cells a prolonged survival in this proliferative region. In the base of the crypt no activity of the tumour suppressor protein APC has been found. In the intermediate portion of the colonic crypt, the activity of survivin is diminished, a fact which correlates with colonocytes not proliferating and starting to differentiate and mature. On the other hand, the greatest activity of APC in this region has been associated with the suppression of survivin, suggesting a possible inhibition of survivin by APC. In the upper portion of the crypt, great activity of the protein APC has been found, while the levels of survivin are low or non-existent.
This region already includes the mature colonocytes that later will suffer terminal apoptosis and be extruded (46).
How could colon carcinogenesis be initiated? A very sensible hypothesis is through inactivation of APC due to a mutation. It has been shown that an APC mutation is the initiating molecular event in colorectal tumorigenesis. When APC, whose function in the colonocyte is to control cell proliferation through its association with ß-catenin, is inactivated, there will be neither differentiation nor maturation of colonocytes, and stem cells proliferating at the base of the crypt will migrate towards the apex without maturation. But on the other hand, mutant APC will not be able to inhibit survivin, thus allowing its constitutive expression, and thereby inhibiting apoptosis and causing an abnormal accumulation of cells at the apex of crypts, which will give rise to adenomas and ultimately tumours. These cells tend to maintain their natural non-differentiated, stem-cell phenotype while they migrate towards the apex of the crypt and continue proliferating (46). This is the hypothesis that explains carcinogenesis via the gatekeeper that is APC.
THE CARETAKERS WAY
There is another possible colorectal carcinogenesis pathway: through caretaker genes. Although this possibility is not as well documented as the previous one, it is another way colorectal carcinogenesis is made possible, giving rise not only to sporadic cancers but also to familial and hereditary forms. There are, at least, six DNA mismatch repair genes, and their implication in the hereditary syndrome HNPCC is well known (47).
Cells must preserve their genome integrity in order to avoid the inheritance of deleterious mutations by daughter cells and the accumulation of mutations in genes that control cell proliferation. If this defence is breached, the result is the growth of malignant tumours (48). Cells employ various DNA repair systems to safeguard the integrity of their genome.
The MMR system eliminates errors in mismatches be-tween bases, and also insertion-deletion loops resulting from the DNA polymerase slipping during replication. The first lesions affect non-repetitive DNA and result in base substitutions (i.e. G«T), while the loops affect repetitive DNA and result in short repetitive units (CA) added or subtracted in microsatellites. This is known as microsatellite instability (MSI) (48). MSI is found both in sporadic and hereditary (HNPCC) colorectal tumours, but it is a distinctive characteristic of HNPCC colorectal tumours. The majority of tumours presenting MSI owe this characteristic to the inactivation of one of the MMR genes: the hMLH1 gene. This inactivation is mainly due to hypermethylation and not to somatic mutations or loss of heterozygosis (49). Nevertheless, germinal mutations associated with HNPCC are mainly mutations in the hMLH1 and hMSH2 repair genes, and their products are critical for MSI to develop (48).
The candidate model of carcinogenesis via caretakers is based on a 100-1000 times increase of the mutation rate compared to normal cells. Besides microsatellites, mutations hit genes that control cell proliferation, specifically those containing repetitive sequences as mutation targets (50). In HNPCC, besides the inherited mutation in the MMR genes, a somatic mutation is required to inactivate both gene copies. Due to the error in the MMR system, errors that normally occur during DNA replication remain uncorrected, and thus mutations accumulate and different regions of the genome become affected, damaging in turn both alleles of tumour suppressor genes, which results in the development of a tumour. The prolonged initiation of this tumour is followed by a fast progression, due to mutation rate acceleration (13,14,16,17).
The probability that an individual with a defective DNA mismatch repair system will develop an adenoma could be not greater than that of the general population, but once an adenoma develops, its progression to cancer is faster in the individual with defective repair genes since the colon environment will induce irreparable damage (4,51).
CONCLUSION
We have seen that colorectal carcinogenesis may start by the inactivation of tumour suppressor genes, whether gatekeepers or caretakers; that this inactivation may result from mutation or from hypermethylation of these genes; that there are other genes that may be hit by activating or inactivating mutations, as oncogenes and genes participating in cell proliferation control and apoptosis; that in all tumours there is accumulation of multiple mutations in the previously mentioned genes, and that all colorectal cancers do not exhibit the same mutations, neither qualitatively nor quantitatively.
Obviously, the colorectal carcinogenesis pathway is not unique, and probably there are various ways for the initiation, development, and progression of colorectal cancer. Not all has been elucidated at present, but every step taken towards the development of colorectal carcinogenesis research will be a step towards an understanding of its mechanisms and a way to prevent this devastating illness.
REFERENCES
1. Burt RW. Colon Cancer Screening. Gastroenterology 2000; 119: 837-53. [ Links ]
2. National Cancer Institute. Cancer genetics Overview. Available at: http: //www.cancer.gov/cancerinfo/pdq/genetics/overview.html Date last modified: 06/2002. [ Links ]
3. Burt RW, DiSario JA, Cannon-Albright LA. Genetics of colon cancer: Impact of inheritance on colon cancer risk. Annu Rev Med 1995; 46: 371-9. [ Links ]
4. Potter, JD. Colorectal cancer: Molecules and population. J Nat Cancer Inst 1999; 91: 916-32. [ Links ]
5. Lynch HT, Lynch JF. Genetics of colorectal cancer. Digestion 1998; 59: 481-92. [ Links ]
6. Online Mendelian Inheritance in Man, OMIM (TM). John Hopkins University, Baltimore, MD. MIM Number: 175100: January 29, 2003: World Wide Web URL: http: //www.ncbi.nlm.nih.gov/omim/. [ Links ]
7. Wu JS, Paul P, Mc Gannon EA, Church JM. APC genotype, polyp number and surgical options in familial adenomatous polyposis. Ann Surg 1998; 227: 57-62. [ Links ]
8. Burt RW, Jacoby RF. Polyposis syndromes. In: Yamalda T, ed. Text book of gastroenterology. 3rd. ed. Philadelphia: Lippincott Raven, 1999. p. 1995-2022. [ Links ]
9. Guillem JG, Smith AJ, Culle J, Ruo L. Gastrointestinal polyposis syndromes. Curr Prob Surg 1999; 36: 219-323. [ Links ]
10. King JE, Dozois RR, Lindor NM, Alquist DA. Care of patients and their families with familial adenomatous polyposis. May Clin Proc 2000; 75: 57-67. [ Links ]
11. Vasen RFA, Mecklin JP, Meera-Khan P, Lynch HT. International Collaborative Group on hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Dis Colon Rectum 1991; 34: 424. [ Links ]
12. Rodríguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997; 89: 1758-62. [ Links ]
13. N Katballe, M Christensen, F P Wikman, T F Ørntoft, S Laurberg. Frequency of hereditary non-polyposis colorectal cancer in Danish colorectal cancer patients. Gut 2002; 50: 43-51. [ Links ]
14. Hahn M, Saeger HD, Schackert HK. Hereditary colorectal cancer: clinical consequences of predictive molecular testing. Int J Colorectal Dis 1999; 14: 184-93. [ Links ]
15. Mecklin JP, Peltomaki P. Genetic changes associated with colon tumor development. Ann Chiru Gynaecol 2000; 89: 211-5. [ Links ]
16. Carethers JM. The cellular and molecular pathogenesis of colorectal cancer. Gastroent Clinics North Am 1996; 25: 737-54. [ Links ]
17. Kinzler KW, Vogelstein B. Cancer susceptibility genes. Gatekeepers and Caretakers. Nature 1997; 386: 761-3. [ Links ]
18. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli. Cell 1991; 66: 589-600. [ Links ]
19. Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991; 253: 665-9. [ Links ]
20. Thliveris A, Albertsen H, Tuohy T, Carlson M, Groden J, Joslyn G, et al. Long-range physical map and deletion characterization of the 1100-kb Not I restriction fragment harboring the APC gene. Genomics 1996; 34: 268-70. [ Links ]
21. Horii A, Nakatsuru S, Ichii S, Nagase H, Nakamura Y. Multiple forms of the APC gene transcripts and their tissue-specific expression. Hum Mol Genet 1993; 2: 283-7. [ Links ]
22. Beroud C, Soussi T. APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 1996; 24: 121-4. [ Links ]
23. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet 2001; 10: 721-33. [ Links ]
24. Su LK, Jonson KA, Smith KJ, Hill DE, Vogelstein B, Kinzler KW. Association between wild-type and mutant APC gene products. Cancer Res 1993; 53: 2728-31. [ Links ]
25. Huelsken J, Birchmeier W. New aspects of Wnt signaling pathways in higher vertebrates. Curr Opin Genet Dev 2001; 11: 547-53. [ Links ]
26. Fodde R. The multiple functions of tumour suppressors: it's all in APC. Nat Cell Biol 2003. [ Links ]
27. McEwen DG. Wnt signaling: The naked truth? Curr Biol 2001; 11: R524-R526. [ Links ]
28. van Es JH, Giles RH, Clevers HC. The many faces of the tumor supresor gene APC. Exp Cell Res 2001; 264: 126-34. [ Links ]
29. Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 1998; 19: 379-83. [ Links ]
30. Fuchs E, Segre JA. Stem cells: a new lease on life. Cell 2000; 100: 143-55. [ Links ]
31. Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol 2003; 129: 199-221. [ Links ]
32. Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem 1997; 272: 24735-8. [ Links ]
33. Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev 1999; 9: 15-21. [ Links ]
34. Kobayashi M, Honma T, Matsuda Y, Suzuki Y, Narisawa R, Ajioka Y, et al. Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer 2000; 82: 1689-93. [ Links ]
35. Rubinfeld B, Albert I, Porfiri, E, Munemitsu S, Polakis P. Loss of beta-catenin regulacion by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res 1997; 57: 4624-30. [ Links ]
36. Roose J, Clevers H. TCF transcription factors: molecular switches in carcinogenesis. Biochim Biophys Acta 1999; 1424: M23-M37. [ Links ]
37. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science 1998; 281: 1509-12. [ Links ]
38. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422-6. [ Links ]
39. Gumbiner BM. Signal transduction of beta-catenin. Curr Opin Cell Biol 1995; 7: 634-40. [ Links ]
40. Kintner C. Regulation of embryonic cell adhesion by the cadherin cytoplasmic domain. Cell 1992; 69: 225-36. [ Links ]
41. Mahmoud NN, Boolbol SK, Bilinski RT, Martucci C, Chadburn A, Bertagnolli MM. Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res 1997; 57: 5045-50. [ Links ]
42. Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997; 3: 917-21. [ Links ]
43. Bao R, Connolly DC, Murphy M, Green J, Weinstein JK, Pisarcik DA, et al. Activation of cancer-specific gene expression by the Survivin promoter. J Natl Cancer Inst 2002; 94: 522-8. [ Links ]
44. Altieri DC, Marchisio PC, Marchisio C. Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab Investg 1999; 79: 1327-33. [ Links ]
45. Altieri D. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol Med 2001; 7 (12): 542-7. [ Links ]
46. Zhang T, Otevrel T, Gao Z, Gao Zh, Ehrlich SM, Fields JZ, et al. Evidence that TPC regulates Survivin expression: A possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res 2001; 62: 8664-7. [ Links ]
47. Online Mendelian Inheritance in Man, OMIM (TM). John Hopkins University, Baltimore, MD. MIM Number: 120435. November 16, 2000: World Wide Web URL: http: //www.ncbi.nlm.nih.gov/omim/. [ Links ]
48. Levitt NC, Hickson ID. Caretaker tumour suppressor genes that defend genome integrity. Trends in Molecular Medicine 2002; 8: 179-86. [ Links ]
49. Kuismanen SA, Holmberg MT, Salovaara R, de la Chapelle A, Peltomaki P. Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am J Pathol 2000; 156: 1773-9. [ Links ]
50. Bhattacharyya NP, Skandalis A, Ganesh A, Groden J, Meuth M. Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci USA 1994; 91: 6319-23. [ Links ]
51. Hardy RG, Meltzer SJ, Jankowski JA. ABC of colorectal cancer. Molecular basis for risk factors. BMJ 2000; 321: 886-9. [ Links ]

