










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.8 Madrid ago. 2004
|
TRABAJOS ORIGINALES |
Lesiones pulmonares y niveles de citoquinas tras la administración intraperitoneal
de homogeneizado pancreático en ratas
G. Mozo, M. L. del Olmo, A. Caro-Patón, E. Reyes1, L. Manzano1, A. Belmonte, A. Almaraz y M. Álvarez-Mon1
Departamento de Medicina. Facultad de Medicina. Universidad de Valladolid. 1Departamento de Inmunología.
Facultad de Medicina. Universidad de Alcalá de Henares. Madrid
RESUMEN
Introducción: nuestro objetivo es investigar, en animales sanos, los efectos de la administración de homogeneizado pancreático, con y sin activación enzimática, sobre los niveles séricos de citoquinas y el desarrollo de lesiones pulmonares.
Material y métodos: se estudiaron 106 ratas Wistar macho divididas en 3 grupos: A: administración intraperitoneal de homogeneizado pancreático activado con enteroquinasa; B: homogeneizado sin enteroquinasa; y C: control, con la administración de suero fisiológico. Cada grupo fue dividido en 4 subgrupos de acuerdo al tiempo de sacrificio: 0, 2, 6 y 24 horas. Estudiamos la histología pancreática y pulmonar, parámetros séricos de función renal, hepática y los niveles séricos de IL-1ß, IL-6 y TNFa.
Resultados: no hubo mortalidad en ningún grupo. Se observaron alteraciones pancreáticas en los grupos A y B a las 24 horas. Estos dos grupos presentaron niveles de transaminasas significativamente más elevados que aquellos del grupo control. Los niveles séricos de creatinina estaban más elevados en el grupo A. Los valores de IL-1ß fueron significativamente más altos a las 6 horas en ambos grupos A y B, aunque mayor en el grupo A, el cual también presentó más lesiones histológicas pulmonares en relación con los controles a las 6 horas.
Conclusiones: los niveles de IL-1ß más elevados en el grupo A podrían deberse a la activación enzimática del homogeneizado que podría inducir la producción de esta citoquina por los macrófagos peritoneales, lo que a su vez podría ser el promotor de las lesiones pulmonares.
Palabras clave: Páncreas. Pancreatitis. Interleucina 1ß. TNFa. Pulmón.
INTRODUCCIÓN
En el curso de una pancreatitis aguda grave, el proceso inflamatorio que inicialmente está localizado y limitado al páncreas puede extenderse, inducir una respuesta inflamatoria sistémica, la cual puede progresar hasta causar un fallo multiorgánico (FMO) (1-3). En estos casos hay una acumulación de leucocitos, preferentemente polimorfonucleares (PMN) en varios órganos, tales como el riñón, el hígado y el pulmón, que es paralelo a la gravedad de la PA (4-6). Los PMN se adhieren a las células endoteliales vasculares y su subsiguiente extravasación e infiltración parenquimatosa está mediada por las citoquinas proinflamatorias, tales como el TNFa, la IL-1ß, la IL-6 y la IL-8. Estas citoquinas son inicialmente producidas por las células acinares y los tejidos peripancreáticos lesionados y más tarde producidos por otros órganos (3,7-12). Muchos trabajos han estudiado en profundidad la contribución que la ascitis pancreática pudiera tener en la patofisiología, desarrollo y degeneración de la PA y en la afectación sistémica (13-23). Se ha demostrado que durante la PA hay un incremento en la permeabilidad peritoneal (24,25) que contribuye al paso de las enzimas pancreáticas a través del peritoneo al conducto torácico (26). Además, en la ascitis pancreática se detectan citoquinas proinflamatorias, cuyo origen parece estar en los macrófagos peritoneales activados (15,19,20,27), que contribuyen a la generalización de la inflamación. Sin embargo, estas citoquinas no parecen ser los únicos factores inflamatorios presentes en la ascitis, ya que esta última continúa provocando efectos sistémicos aun cuando esté libre de ellas (15,17-19,28-33).
El lavado peritoneal se ha propuesto como una medida para eliminar los factores proinflamatorios (34-36) presentes en la ascitis pancreática, ya que contribuirían al deterioro de la enfermedad (1), produciendo alteraciones hemodinámicas y en órganos a distancia tales como el riñón, el hígado y el pulmón (28,31,37-42). En el pulmón pudiera inducir el síndrome de distrés respiratorio del adulto (SDRA), como lo indican estudios experimentales cuando se inyecta este exudado en la arteria pulmonar (33) o por vía intravenosa (17) o intraperitoneal (i.p.) (24).
Estudios experimentales han empleado la administración i.p. de homogeneizado pancreático como un método que simula la ascitis pancreática (43). Parece que este homogeneizado, una vez inyectado en una rata sana, adquiere propiedades fibrinolíticas similares a las presentes en la ascitis pancreática que inicialmente no poseía. Ello parece indicar que el homogeneizado interacciona con el peritoneo (43).
El objetivo de este trabajo es estudiar los efectos que en animales sanos provoca la administración de un homogeneizado pancreático con o sin activación previa con enteroquinasa, tanto en los niveles de citoquinas séricas como en el desarrollo de alteraciones en la morfología pulmonar.
MATERIAL Y MÉTODOS
Se han estudiado un total de 106 ratas Wistar macho. El proceso fue efectuado de acuerdo a las normas establecidas por el Comité Ético de la Facultad de Medicina de la Universidad de Valladolid, las cuales siguen las recomendaciones estipuladas en el Decreto 223/1988 de 14 de marzo y en la Orden de 13 de octubre de 1989. Los animales permanecieron en jaulas de 5 individuos con ciclos alternantes de 12 horas de luz y oscuridad, acceso libre a una comida estándar y agua hasta 12 horas antes del comienzo del experimento, en que se les mantuvo en ayuno aunque con libre acceso al agua.
Las ratas fueron distribuidas en 3 grupos:
Grupo A. 28 animales a los que se les administró homogeneizado pancreático previamente activado con enteroquinasa (EK).
Grupo B. 28 animales con administración de homogeneizado pancreático sin previa activación con EK.
Grupo C. 50 animales control que recibieron suero fisiológico.
Cada grupo fue dividido en subgrupos considerando el momento del sacrificio del animal en: 0, 2, 6 y 24 horas a partir del momento de la administración de homogeneizado.
Preparación y administración del homogeneizado pancreático
Se utilizaron 8 ratas Wistar hembra. Fueron dejadas en ayuno durante 12 horas antes del sacrificio pero con libre acceso al agua. Tras la anestesia con Pentothal sódico (B. Braun Medical, Barcelona) i.p. a dosis de 25 mg, se procedió a la laparotomía y extracción del páncreas siguiendo una técnica aséptica. El tejido fue inmediatamente suspendido en una solución estéril de Tris buffer, pH 7.8, con Triton X 100. El páncreas fue homogeneizado en frío y centrifugado a una temperatura de 4 ºC. Cada 20 ml del sobrenadante fue incubado durante 30 minutos a 37 ºC con 2,22 ml de EK (Sigma, Londres) (5 mg/ml en tampón Tris buffer, pH 7,8) que fue utilizado en los animales del grupo A. En el grupo B el homogeneizado fue incubado sin EK.
Se comprobó la actividad tripsínica de este homogeneizado (120 UI/g proteína) para el grupo A mediante el método de Erlanger-Kokowsky-Cohen (44), que estuvo ausente en el no incubado con EK. También se observó previamente que la inyección i.p. de 1 ml de Triton X 100 al 1/1000 más 1 ml de suero fisiológico en la rata no producía ningún tipo de alteración.
Los homogeneizados fueron inyectados i.p. a dosis de 0,5 ml a cada animal, y este momento fue considerado el tiempo 0.
Una vez transcurridos los distintos tiempos, los animales fueron sacrificados siguiendo un protocolo previamente establecido con exsanguinación por punción cardiaca, previa anestesia con 25 mg de Pentothal sódico inyectado i.p. Posteriormente, se realizó una toracotomía y se procedió a extraer el pulmón derecho y una laparotomía media amplia para extraer el páncreas.
Determinaciones bioquímicas y de citoquinas
La sangre obtenida mediante punción cardiaca fue mantenida en hielo durante 90 minutos hasta la formación de coágulo. Tras la centrifugación, el suero fue congelado a -20 ºC hasta su posterior utilización en las determinaciones bioquímicas y a -80 ºC para la determinación de citoquinas.
En el suero se determinaron los niveles de amilasa, lipasa, urea, creatinina, AST, ALT y glucosa utilizando un autoanalizador automático Hitachi 917. El valor del hematocrito fue determinado por centrifugación capilar utilizando sangre heparinizada.
De igual forma, el TNFa, la IL-1ß y la IL-6 fueron determinadas utilizando "kits" comerciales de ELISA para rata y para cada citoquina. Se utilizó un "kit" con una sensibilidad de 20-1000 pg/ml (Diaclone, France) para el TNFa, uno con una sensibilidad de 10-1500 pg/ml (Endogen, USA) para la IL-6 y uno con una sensibilidad entre 3-2000 pg/ml (Biosurce Internacional, USA) para la IL-1ß. La lectura se efectuó con un espectrofotómetro Delta Soft versión 4.1 (Biometallics. Inc) con longitudes de onda de 450 nm con un ordenador Macintosh LC II.
Estudio morfológico
El pulmón derecho y el páncreas de cada animal se pesaron en fresco y fueron sumergidos en una solución de formaldehído al 10%, siendo posteriormente teñidos con hematoxilina/eosina. Ambos órganos fueron estudiados con un microscopio óptico (Zeiss II, Inc, New York) con 200, 400 y 1.000 aumentos. Los parámetros valorados en cada órgano fueron graduados de acuerdo a una escala de 0 a 3 como se muestra en la tabla I.

Se realizó también una evaluación de la histología pulmonar y pancreática calculando la media de un amplio número de parámetros histológicos de cada uno de estos órganos siguiendo un método ya establecido por otros autores (45,46).
La relación del peso de cada órgano en fresco con el peso corporal de cada animal fue dada en porcentaje y fue usada como una estimación del edema tisular (pulmón y páncreas).
Análisis estadístico
Los resultados se expresaron como media ± error estándar de la media (esm). Utilizamos un programa estadístico SPSS 6.1.2. Empleamos la U de Mann Whitney como prueba no paramétrica para la comparación de medias y tomamos una p≤ 0,05 como valor estadísticamente significativo.
RESULTADOS
No hubo mortalidad en ninguno de los grupos estudiados.
Niveles enzimáticos en ambos homogeneizados
Los niveles de lipasa y amilasa en el homogeneizado sin activación con EK fueron más elevados que aquellos del homogeneizado activado (lipasa 122,1 KU/ml contra 97,2 KU/ml, amilasa 298,7 U/ml contra 258,3 U/ml).
Los niveles de transaminasas también fueron más elevados en el homogeneizado sin activar con EK que en el homogeneizado activado (AST 6430 mU/ml contra 3450 mU/ml, ALT 2135 mU/ml contra 312 mU/ml).
Los resultados de la estimación de proteínas demostraron una mayor concentración en el homogeneizado sin activar con EK que en el activado (1,81 g/dl vs 1,13 g/dl).
Evaluación pancreática
No hubo alteraciones pancreáticas macroscópicas en los grupos A y B en comparación con el control, ni hubo diferencias en la relación peso pancreático/peso corporal.
Se observaron niveles de amilasa sérica significativamente más elevados a las 2 y 6 horas en el grupo B con respecto al control. La lipasa sérica mostró valores ligeramente más elevados en ambos grupos (A y B) a las 2 y 6 horas, que fueron estadísticamente significativos con respecto al control (Tabla II).

Cuando se compararon los grupos experimentales, observamos que el grupo B tenía niveles de amilasa a las 6 horas significativamente más altos que el grupo A. Esto mismo también sucedió en relación con la lipasa en los tiempos 2 y 6 horas (Tabla II).
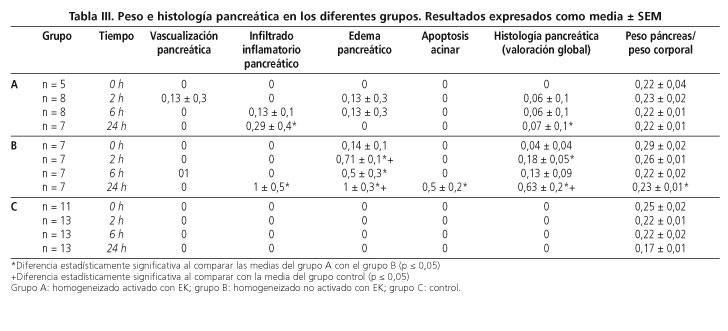
El estudio histológico del páncreas demostró la presencia de infiltración inflamatoria acinar y edema pancreático a las 24 horas en el grupo A (con EK), una diferencia que fue significativa en relación con el grupo control. En el grupo con homogeneizado sin EK (grupo B), se observó a las 24 horas una infiltración inflamatoria de los acini y ligera apoptosis, mientras que a las 2, 6 y 24 horas se encontró edema acinar. En todos los tiempos, estos cambios tenían significación estadística en relación con los controles y a las 2 y 24 horas con respecto al grupo A (Tabla III).
Transaminasas séricas y parámetros de función renal
Ambos grupos experimentales presentaron a las 2 y 6 horas niveles séricos de AST significativamente más elevados que el grupo control. En esos mismos tiempos, los niveles séricos de ALT fueron más elevados en el grupo B con respecto a las controles y en relación con el grupo A (Tabla II).
No hubo diferencias en los valores de urea sérica entre los grupos. La creatinina sérica presentó en el grupo A unos niveles significativamente más elevados que en los controles a las 6 horas. No hubo diferencias en la glucosa ni en el hematocrito (Tabla II).
Niveles séricos de IL-1ß, TNFa e IL-6 (Tabla II)
La determinación de la IL-1ß mostró niveles significativamente más elevados a las 6 horas en ambos grupos A y B comparados con el grupo control, apareciendo los valores más altos en el grupo A (58,8 pg/ml) en contraste con el grupo B (22,4 pg/ml) pero sin que las diferencias alcanzaran significación estadística. Lo mismo ocurrió a las 0 horas en el grupo B (Tabla II).
La determinación de la IL-6 no mostró cambios significativos en ningún grupo motivo por lo que no se incluyen estos datos en la tabla II.
Los resultados del TNFa tampoco mostraron diferencias estadísticamente significativas cuando se compararon los dos grupos experimentales entre sí o con el grupo control durante los diferentes tiempos del experimento.
Alteraciones en la histología pulmonar
En el grupo A, observamos una mayor alteración de la arquitectura a las 6 horas en comparación con el grupo control y con el grupo B, junto con una mayor presencia de edema pulmonar a nivel alveolo-capilar en el mismo periodo. Estas diferencias eran estadísticamente significativas. En el grupo B, no se observaron alteraciones en relación con el control en ninguno de los parámetros analizados (Tabla IV), Fig. 1).

La valoración de la relación peso del pulmón derecho/peso corporal no mostró diferencias significativas entre los grupos.
DISCUSIÓN
De acuerdo con nuestro estudio, y como era de esperar, no observamos alteraciones estructurales pancreáticas significativas en los grupos experimentales. La presencia de un infiltrado inflamatorio muy discreto y de un mínimo edema intersticial en ambos grupos a las 24 horas podría estar relacionado con una agresión tardía al páncreas, probablemente de origen inmunológico. Se puede especular que la administración de un homogeneizado pancreático proveniente de otro animal pudiera generar una respuesta inmune contra la propia víscera. Una posibilidad más remota, sin embargo, pudiera ser que el incremento de la IL-1ß producida en ambos grupos a las 6 horas provocase una quimiotaxis de células inflamatorias selectiva y tardía sobre le páncreas. En cualquier caso, estas alteraciones son tan escasas que no parece que hayan influido ni en la afectación pulmonar ni en la producción de citoquinas, ya que estos fenómenos aparecen en fases más tempranas.
Dada la ausencia de lesión pancreática, los incrementos en la lipasa y amilasa séricas podrían ser debidos a absorción peritoneal de los enzimas contenidos en el homogeneizado administrado, hecho que también se produce en la propia ascitis pancreática (47,48). Las diferencias en los niveles de lipasa y amilasa entre los grupos A y B, más elevadas en B, concuerdan con la mayor actividad de lipasa y amilasa observada en el homogeneizado no activado con EK. Se pudiera pensar que los niveles enzimáticos menores en el homogeneizado activado con EK podrían ser el resultado de la digestión por la tripsina presente en él.
Igualmente, los valores mas elevados de ALT a las 2 y 6 horas en el grupo B pudieran también ser interpretados de esta misma manera. Sin embargo, varios estudios han demostrado la presencia de apoptosis de células hepáticas en animales sanos después de la administración i.p. de ascitis pancreática (42). En este sentido, el homogeneizado pancreático utilizado en nuestro estudio pudiera reproducir este mismo efecto.
La ausencia de niveles séricos detectables de IL-6 y de diferencias significativas en los valores de TNFa pudieran explicarse por el hecho de que las lesiones producidas no fueran de suficiente intensidad, como ya ha sido puesto de relieve en otros trabajos experimentales o en humanos (39,49,50).
Los niveles de IL-1ß a las 6 horas permanecieron significativamente más altos en ambos grupos de animales en relación con el grupo control. Como el homogeneizado pancreático provenía de ratas sanas, sin inflamación pancreática, no parece ser este el origen de la citoquina. Aunque nosotros no hemos estudiado histológicamente el peritoneo, se han publicado estudios que usan modelos similares al nuestro en los que no pudieron detectar la presencia de signos inflamatorios peritoneales (31). Por ello, podemos pensar que la elevación de IL-1ß en los grupos A y B no era debida a la presencia de una inflamación peritoneal. Tampoco parece que el páncreas o el pulmón sean el origen probable de esta citoquina ya que no existe lesión en estos órganos previa al incremento de la IL-1ß. Así pues, sugerimos que el origen más probable de esta citoquina pudiera encontrarse en los macrófagos intraperitoneales. Numerosos trabajos han demostrado la producción de citoquinas pro-inflamatorias y un aumento de proteínas reguladoras de la trascripción de genes pro-inflamatorios, tales como NκB por los macrófagos peritoneales y esplénicos tras ser estimulados con ascitis pancreática carente de endotoxinas y desprovista de citoquinas (15,18-20,28-33).
Si tenemos en cuenta que los enzimas pancreáticos liberados a la circulación no parecen influir sobre la producción de citoquinas por los macrófagos (19,20), ha de existir un factor de origen pancreático presente en el homogeneizado, no dependiente de la existencia de una lesión pancreática previa, que actúe sobre los macrófagos peritoneales y los estimule a producir IL-1ß.
El hecho de que en el grupo A los niveles de IL-1ß a las 6 horas sea mayor que en el grupo B, y que sólo en el grupo A se hayan producido lesiones pulmonares significativas sugiere que este factor desconocido se encuentre en mayor cantidad en el homogeneizado activado (¿un producto de la transformación por los enzimas activados de alguna proteína pancreática?). Otra posibilidad pudiera ser que la presencia de enzimas activados contribuya al efecto de este factor sobre los macrófagos peritoneales, y aunque parece que los enzimas no son directamente responsables de estos fenómenos, es probable que los favorezcan (18,20,21,23).
En cuanto al estudio de la morfología pulmonar, en el grupo A, se apreciaron alteraciones en la estructura pulmonar normal y la presencia de un mínimo edema pulmonar, estadísticamente significativo a las 6 horas en comparación con los grupos B y C. La ausencia de diferencias en el peso del pulmón puede ser debida al bajo grado de edema con ausencia de hallazgos macroscópicos junto con un error de muestreo, ya que el pulmón derecho extirpado a los animales no tenían exactamente del mismo tamaño en todos los animales, y no siempre existe un claro paralelismo entre el tamaño pulmonar y el peso corporal.
Es difícil establecer el papel que los enzimas activados del homogeneizado pancreático pudieran haber jugado en la generación de las lesiones pulmonares. Un reciente estudio ha demostrado una mayor lesión pulmonar asociada a PA cuando se administra EK intravenosa con el fin de activar los enzimas pancreáticos que se encuentren fuera de los acinis. Sin embargo, este procedimiento suele causar un agravamiento de la lesión pancreática, por lo que el incremento de la lesión pulmonar debe atribuirse más a la severidad de la PA que a la propia actividad de las enzimas pancreáticas sobre el pulmón (51). Teniendo en cuenta que los enzimas activados por sí solos no provocan lesión pulmonar (18) y que tanto en la PA como en otras enfermedades es patente la estrecha relación existente entre la presencia de citoquinas pro-inflamatorias y la aparición de lesión pulmonar (52-55,56), pudiéramos relacionar las alteraciones pulmonares presentes en el grupo A con los niveles más elevados de IL-1ß en sangre que vemos en nuestros animales. No obstante, a las 2 horas, la IL-1ß está más elevada en el grupo B que en el grupo A, aunque esta diferencia no es significativa, lo cual hace dudar del papel exclusivo de la IL-1ß en la producción de las lesiones pulmonares. En este sentido, pudiera haber algún otro factor presente en el homogeneizado activado por EK que contribuya a la aparición de esas lesiones.
En conclusión, el hecho de que sólo se observaran lesiones pulmonares en animales tratados con homogeneizado activado donde los niveles de IL-1ß estuvieron significativamente más elevados en relación con el control sugiere que los enzimas pancreáticos pudieran ser posibles facilitadores de una interacción entre el homogeneizado activado y los macrófagos peritoneales. En el caso de una verdadera ascitis pancreática, todos estos elementos junto con la inflamación pancreática, podrían converger generando, indudablemente, mayores niveles de citoquinas pro-inflamatorias y alteraciones sistémicas.
BIBLIOGRAFÍA
1. Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg 1998; 175: 76-83.7. [ Links ]
2. Ogawa M. Acute pancreatitis and cytokines: "Second Attack" by septic complication leads to organ failure. Pancreas 1998; 16: 312-5. [ Links ]
3. Sakorafas GH, Tsiotou AG. Etiology and pathogenesis of acute pancreatitis: current concepts. J Clin Gastroenterol 2000; 30: 343-56. [ Links ]
4. Werner J, Dragotakes SC, Fernández-del Castillo C, Rivera JA, Ou J, Rattner DW, et al. Technetium-99m-labeled white blood cells. A new method to define the local and systemic role of leukocytes in acute experimental pancreatitis. Ann Surg 1998; 227: 86-94. [ Links ]
5. Werner J, Z'graggen K, Fernández-del Castillo C, Lewndrowski KB, Compton CC, Warshaw AL. Specific therapy for local and systemic complications of acute pancreatitis with monoclonal antibodies against ICAM- 1. Ann Surg 1999; 226: 834-42. [ Links ]
6. O'Neill S, O'Neill AJ, Conroy E, Brady HR, Fitzpatrick JM, Watson RW. Altered caspase expression results in delayed neutrophil apoptosis in acute pancreatitis. J Leukoc Bil 2000; 68: 15-20. [ Links ]
7. Osman MO, Lausten SB, Jakobsen NO, Kristensen JU, Deleuran B, Larsen CG, et al. Graded experimental acute pancreatitis monitoring of a renewed rabbit model focusing on the production of interleukin-8 (IL-8) and CDI lb/CDI8. Eur J Gastroenterol Hepatol 1999; 11: 137-49. [ Links ]
8. Gukovskaya A, Gukosky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, et al. Pancreatic acinar cells produce, release and respond to Tumor Necrosis Factor-a. Role in regulating cell death and pancreatitis. J Clin Invest 1997; 100: 1853-62. [ Links ]
9. Chen C-C, Wang S-S, Lu R-H, Chang F-Y, Lee S-D. Serum interleukin 10 and interleukin 11 in patients with acute pancreatitis. Gut 1999; 45: 895-9. [ Links ]
10. Vaccaro MI, Ropolo AA, Grasso D, Calvo EL, Rerreira M, Iovanna JL, et al. Pancreatic acinar cells submitted to stress activate TNF-alpha gene expression. Biochem Biophys Res Commun 2000; 268: 485-90. [ Links ]
11. Norman JG, Fink GW, Denham W, Yang J, Carter G, Sexton CH, et al. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for Distant Organ Dysfunction. Dig Dis Sci 1997; 42: 1783-88. [ Links ]
12. Kim H, Seo JY, Kim KH. NF-kappaB and cytokines in pancreatic acinar cells. J Korean Med Sci 2000; 15 (Supl. 1): S53-4. [ Links ]
13. Masamune A, Shitnosegawa T, Kimura K, Fujita M, Sato A, Koizumi M, et al. Specific induction of adhesión molecules in human vascular endothelial cells by rat experimental pancreatitis-associated ascitic fluids. Pancreas 1999; 18: 141-50. [ Links ]
14. Takase K, Takeyama Y, Nishikawa J, Ueda T, Hori Y, Yamamoto M, et al. Apoptotic cell death of renal tubules in experimental severe acute pancreatitis. Surgery 1999; 125: 411-20. [ Links ]
15. Satoh A, Shimosegawa T, Masamune A, Fujita M, Koi M, Toyota T. Ascitic fluid of experimental severe acute pancreatitis modulases the function of peritoneal macrophages. Pancreas 1999; 19: 268-75. [ Links ]
16. Hori Y, Takeyalna Y, Ueda T, Shinkai M, Takase K, Kuroda Y. Macrophage-derived transforming growth factor-beta 1 induces hepatocellular injuty via apoptosis in rat severe acute pancreatitis. Surgery 2000; 127: 641-9. [ Links ]
17. Denham W, Jun Y, Wang H, Botchkina G, Tracey KJ, Norman J. Inhibition of p38 mitogen activate kinase attenuates the severity of pancreatitis-induced adult respiratory distress syndrome. Crit Care Med 2000; 28: 2567-72. [ Links ]
18. Walsh CJ, Leeper-Woodford SK, Carey PD. CD18 adhesion receptors, tumor necrosis factor and neutropenia during septic lung injury. J Surg Research 1991; 50: 323-9. [ Links ]
19. Denham W, Yang J, Norman J. Evidence for an unknown component of pancreatic ascites that induces adult respiratory distress syndrome through an interleukin-1 and tumor necrosis factor-dependent mechanism. Surgery 1997; 122: 295-302. [ Links ]
20. Denhain W, Yang J, Fink G, Zervos EE, Carter G, Norman J. Pancreatic ascites as a powerful inducer of inflammatory cytokines. The role of known vs unknown factors. Arch Surg 1997; 132: 1231-6. [ Links ]
21. Lundberg AH, Eubanks JW 3rd, Henry J, Sabek O, Kotb M, Gaber L, et al. Trypsin stimulates production of cytokines from peritoneal macrophages in vitro and in vivo. Pancreas 2000; 21: 41-51. [ Links ]
22. Jaffray C, Yang J, Norman J. Elastase mimics pancreatitis-induced hepatic injury via inflammatory mediators. J Surg Res 2000; 90: 95-101. [ Links ]
23. Jaffray C, Yang J, Carter G, Mendez C, Nonnan J. Pancreatic elastase activases pulmonary nuclear factor kappa B and inhibitory kappa B, mimicking pancreatitis-associated adult respiratory distress syndrome. Surgery 2000; 128: 225-31. [ Links ]
24. Marotta F, Fesce E, Rezakivic I, Chui DH, Suzuki K, Idéo G. Nafamostat Mesilate on the course of acute pancreatitis. Protective effect on peritoneal permeability and relation with supervening pulmonary distress. Int J Pancreatol 1994; 16: 51-9. [ Links ]
25. Sevensson C, Sjödahl R, Tagesson C, Ihse I. Increased peritoneal permeability in acute experimental pancreatitis. Int J Pancreatol 1989; 4: 83-90. [ Links ]
26. Egdahl RH. Mechanism of blood enzyme changes following production of experimental pancreatitis. Ann Surg 1958; 148: 389-400. [ Links ]
27. Marton J, Szasz Z, Nagy Z, Jarmay K, Takacs T, Lonovics J, et al. Beneficial effect of octreotide treatment in acute pancreatitis in rats. Int J Pancreatol 1998; 24: 203-10. [ Links ]
28. Ofstad E. Formation and destruction of plasma kinins during experimental acute hemorrhagic pancreatitis in dogs. Scand J Gastroenterol 1970; 5 (Supl. 5): 9. [ Links ]
29. Ohlsson K, Tegner H. Experimental pancreatitis in the dog. Demonstration of trypsin in ascitic fluid, lymph and plasma. Scand J Gastroenterol 1973; 8: 129. [ Links ]
30. Thal AP, Kobold E, Hollenberg M. The release of vasoactive substances in acute pancreatitis. Am J Surg 1954; 105: 708. [ Links ]
31. Ellison EC, Pappas TN, Johnson JA, Fabri PJ, Carey LC. Demonstration and characterization of the hemo-concentrating effect of ascitic fluid that accumulate during haemorrhagic pancreatitis. J Surg Res 1981; 30: 241-8. [ Links ]
32. Satoh A, Shimosegawa T, Kimura K, Moriizumi S, Masamune A, Koizumi M, et al. Nitric oxide is overproduced by peritoneal macrophages in rat taurocholate pancreatitis: the mechanism of inducible nitric oxide synthase expression. Pancreas 1998; 17: 402-11. [ Links ]
33. Innes J, Frase I, Carey LC. The vasoactive properties of ascitic fluid in acute pancreatitis in a porcine model. Arch Surg 1986; 121: 665-8. [ Links ]
34. Ranson JHC, Spencer FC. Role of peritoneal lavage in severe acute pancreatitis. Ann Surg 1978; 187: 565-75. [ Links ]
35. Stone NN, Fabian TC. Peritoneal dyalisis in the treatment of acute alcoholic pancreatitis. Surg Gynecol Obstet 1980; 150: 878-82. [ Links ]
36. Yokoi H, Naganuma T, Higashiguchi T, Isaji S, Kawarada Y. Prospective study of a protocol for selection of treatment of acute pancreatitis based on scoring of severity. Digestion 1999; 60 (Supl. 1): 14-8. [ Links ]
37. Frey CF, Wong HN, Hickman D, Pullos T. Toxicity of hemorrhagic ascitic fluid associated with hemorrhagic pancreatitis. Arch Surg 1990; 117: 401-4. [ Links ]
38. Ueda T, Takeyama Y, Hori Y, Shinkai M, Takase K, Goshima M, et al. Hepatocyte growth factor increases in injures organs and functions as an organotrophic factor in rats with experimental acute pancreatitis. Pancreas 2000; 20: 84-93. [ Links ]
39. Heath DI, Cruickshank A, Gudgeon M, Jehanli A, Shenkin A, lmrie CW. Role of interleukin-6 in mediating the acute phase protein response and potential as an early means of severity assessment in acute pancreatitis. Gut 1993; 66: 41-5. [ Links ]
40. Coticchia JM, Lessler MA, Carey LC, Gower WR, Mayer AD, McMahon MJ, et al. Peritoneal fluid in human acute pancreatitis blocks hepatic mitochondrial respiration. Surgery 1986; 100: 850-6. [ Links ]
41. Bielecki JW, Clugosz J, Pawlicka E, Gabryelewicz A. The effect of pancreatitis associated ascitic fluid on some functions of rat liver mitochondria: a possible mechanism of the damage to the liver in acute pancreatitis. Int J Pancreatol 1989; 5: 145-56. [ Links ]
42. Takeyama Y, Hori Y, Takase K, Ueda T, Yamamoto M, Kuroda Y. Apoptotic cell death of hepatocytes in rat experimental severe acute pancreatitis. Surgery 2000; 127: 55-64. [ Links ]
43. Etoh Y, Sumi H, Tsushima H, Maruyama M, Mihara H. Fibrinolytic enzymes in ascites during experimental acute pancreatitis in rats. Int J Pancreatol 1992; 12: 127-37. [ Links ]
44. Erlanger BF, Kokowsky N, Coh W. The preparation and properties of two chromogenic substrates of trypsin. Arch Biochem Biophys 1961; 95: 271-8. [ Links ]
45. Norman JG, Franz MG, Fink GS, Messina J, Fabri PJ, Gower WR, et al. Decreased mortality of severe acute pancreatitis after proximal cytokine blockade. Ann Surg 1995; 221: 625-34. [ Links ]
46. Kruse P, Hage E, Lasson A. Proteases and inhibitors in taurocholate-induced acute pancreatitis in rats. Int J Pancreatol 1999; 25: 113-21. [ Links ]
47. Mayer AD, Airey M, Hodgson J, McMahon J. Enzyme transfer from pancreas to plasma during acute pancreatitis. The contribution of ascitic fluid and lymphatic drainage of the pancreas. Gut 1985; 26: 876-81. [ Links ]
48. Waterman NG, Walsky RS. Transperitoneal absorption of amylase in acute experimental pancreatitis. Surg Gynecol Obstet 1970; 131 (4): 729-32. [ Links ]
49. Biffl WL, Moore EE, Moore FA. Interleukin-6 in the injured patients. Marker of injury or mediator of inflammation. Ann Surg 1996; 224: 647-64. [ Links ]
50. Kishimoto T, Akira S, Narazaki M. Interleukin-6 family of cytokines and gp130. Blood 1995; 86: 1243-54. [ Links ]
51. Hartwig W, Werner J, Jiménez R, Z'graggen K, Weimann J, Lewndrowski KB, et al. Trypsin and activation of circulating trypsinogen contribute to pancreatitis-associated lung injury. Am J Physiol 1999; 277 (Gastrointest. Liver Physiol.40): G1008-G1016. [ Links ]
52. Sameshima H, lkei S, Morí K. The role of tumor necrosis factor-a in the aggravation of cerulein-induced pancreatitis in rats. Int J Pancreatol 1993; 14: 107-15. [ Links ]
53. Suter PM, Suter S, Girardin E, Roux-Lombard P, Grau GE, Dayer J-M. High bronchoalveolar levels of Tumor Necrosis Factor and its inhibitors, Interleukin-1, Interferon, and Elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am Rev Respir Dis 1992; 145: 1016-22. [ Links ]
54. Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, et al. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet 1993; 341: 643-47. [ Links ]
55. Montravers P, Chollet-Martin S, Marmuse JP. Lymphatic release of cytokines during acute lung injury complicating severe pancreatitis. Am J Respir Crit Care Med 1995; 152: 1527-33. [ Links ]
56. Mozo G, del Olmo ML, Caro-Patón A, Reyes E, Manzano L, Belmonte A, et al. Afectación pulmonar y niveles de citocinas en un modelo de pancreatitis aguda experimental. Rev Esp Enferm Dig 2002; 94: 53-9. [ Links ]

