










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.10 Madrid oct. 2004
| ORIGINAL PAPERS |
Influence of rofecoxib on experimental colonic carcinogenesis in rats
J. F. Noguera Aguilar, A. Plaza Martínez1, I. Amengual Antich2, J. M. Morón Camis3, C. Tortajada Collado4
and J. J. Pujol Tugores5
IUNICS. General Surgery Unit and Digestive Diseases. Son Llàtzer Hospital. Balearic Islands. 1Vascular Surgery
Department. Dr. Peset University Hospital. Valencia. 2Pathology Department. Manacor Hospital Foundation.
Valencia. 3General and Digestive Tract Surgery. General Surgery Department. Son Dureta University Hospital.
4General Nurse. Son Dureta University Hospital. 5General and Digestive Tract Surgery. Son Llàtzer Hospital.
Balearic Islands. Spain
ABSTRACT
Aim: to investigate the effect of a selective cyclooxigenase-2 (COX-2) inhibitor, rofecoxib, in the prevalence of experimental colon tumors in rats.
Experimental design: experimental study with 35 male Sprague-Dawley rats, divided into four groups: a) control group without experimental manipulation (n = 5); b) pharmacological carcinogenesis with 1-2 dimethylhydrazine dihydrocloride (n = 10); c) pharmacological carcinogenesis and addition of acetylsalicylic acid (AAS) (n = 10); and d) carcinogenesis and addition of rofecoxib (n = 10). Carcinogenesis was induced with 1-2 dimethylhydrazine at a weekly dose of 25 mg/kg for 18 weeks. Colon tumors were isolated at 20 weeks. Antiinflammatory agents were given at a dose of AAS 30 mg/kg and rofecoxib at 3 mg/kg.
Results: the percentage of colonic tumors was significantly reduced in the rofecoxib group. This result was found for all tumors and for the malignant lesions, adenocarcinomas.
Conclusions: rofecoxib, a selective COX-2 inhibitor, reduced the percentage of drug-induced neoplastic glandular tissue in rats.
Key words: Rofecoxib. Colorectal cancer. Rat. Adenocarcinoma. AAS. Cyclooxigenase.
Noguera Aguilar JF, Plaza Martínez A, Amengual Antich I, Morón Camis JM, Tortajada Collado C, Pujol Tugores JJ. Influence of rofecoxib on experimental colonic carcinogenesis in rats. Rev Esp Enferm Dig 2004; 96: 678-686.
Recibido: 24-09-03.
Aceptado: 17-02-04.
Correspondencia: José Francisco Noguera Aguilar. Avenida Antonio Maura, 91, bajo 11. 07009 Pont d'Inca. Baleares. Fax: 971 202 173. e-mail:jnoguera@hsll.es.
INTRODUCTION
Colorectal cancer (CRC) is the most common digestive cancer in Western Europe and in the United States (1), and the second leading cause of death in the Western world (2). In our country, it has an incidence of 20-30 cases per 100,000 inhabitants per year, and is responsible for 20% of malignancy-induced deaths (3,4). Colorectal cancer has an overall 5-year survival of 60% (5).
There are two major strategies for the prevention of CRC: either early detection of the disease and of premalignant lesions or chemical prevention. Early management of premalignant or early malignant lesions diminishes the incidence of CRC and its related mortality (6-8). Chemical prevention is based on using chemical (pharmacological or otherwise) agents to prevent the development of the carcinogenic process. Various agents have been proposed as possible chemopreventives of CRC, among them drugs such as cyclooxigenase-2 (COX-2) inhibitors.
Cyclooxigenase (COX) is the enzyme catalyzing the first two steps in the synthesis of prostaglandins from arachidonic acid. Two COX isoforms have been identified that are structurally similar and catalyze the same chemical reaction, but acting on different places and times: COX-1 is constituent and always present in all tissues, while COX-2 is inducible by a number of stimuli, mostly in association with both acute and chronic inflammation (9-11).
The activity of COX-2 seems to be related to tumor cell proliferation in aberrant colonic crypts by inhibiting cellular apoptosis -or programmed cell death- and the function of NK (natural killer) T cells, as well as by favoring tumor expansion through the induction of angiogenesis within the tumor (12). COX-2 is inducible by oncogens ras and scr, interleukin-1, hypoxia, benzopyrene, ultraviolet light, epidermal growth factor, and transforming growth factor alpha. Dexametasone, antioxidants, and protein p53 suppress the expression of COX-2. The isoenzyme COX-2 synthesizes prostaglandin E2 (PGE2), which inhibits the apoptosis and associates with the development of metastatic disease. In addition, it induces interleukin-6 (IL-6), which bears some relation to cancer cell invasion, and haptoglobin, which induces angiogenesis and the set-up of these cells.
COX-2 overexpression has been reported in colon tumors; consequently, a specific inhibition of COX-2 should prove a preventive manoeuvre. Many studies have isolated the enzyme COX-2 from the stroma of adenomas and from the stroma and epithelium of CRC (13-15). Likewise, some authors have even related the extent of COX-2 expression to CRC survival (16).
The present study attempts to evaluate whether rofecoxib (a selective COX-2 inhibitor with minimal effect on the COX-1 isoenzyme) has any inhibitory effect on drug-induced carcinogenesis in rats. As a secondary aim of this study was to assess whether this inhibitory effect is greater in rofecoxib treated rats than in the acetylsalicylic acid (AAS) -a non-selective COX inhibitor- group of animals.
Drug-induced carcinogenesis will clarify the protective effect of rofecoxib and AAS on induced tumor development. Carcinogenesis with 1-2 dimethylhydrazine dihydrocloride (DMH) induces aberrant crypt formation at the intestinal epithelium and promotes carcinogenesis after induction. This dysplastic epithelium overexpresses COX-2, therefore a specific inhibition of this isoenzyme may have a suppressing effect on colonic induced tumors.
MATERIAL AND METHODS
Fifty-seven colonic tumors were evaluated in thirty-five male Sprague-Dawley rats (Criffa, Spain). Rats (mean weight of 225 g; range, 165-300 g) were divided into four groups: a) five animals formed the strict control group, without any experimental manipulation, and ten animals in each experimental group; b) a group with drug-induced carcinogenesis (n = 10); c) a group with AAS modulation (n = 10); and d) a group with rofecoxib modulation (n = 10). The carcinogenic induction of colon neoplasms was carried out with DMH (Sigma-Aldrich, Spain). The simultaneous administration of nonsteroid antiinflammatory drugs (NSAIDs) was based on the oral administration of 30 mg/kg AAS (Aspirina®, Bayer, Spain) or 3 mg/kg rofecoxib (Vioxx®, MSD, Spain), both dissolved in the drinking water.
Dietary and environmental conditions
Rats were maintained in type III cages (Criffa, España) at two animals per cage. Environmental conditions at the animal storage area were: 12 h/12 h light/darkness cycle (light from 8:00 am to 8:00 pm); uniform temperature of 22 ± 2 ºC, and relative humidity of 60-70%. Diet provided was maintenance diet A.04 (Panlab, Spain).
The study complied with the guidelines established in Spanish Royal Decree 223/1998 and European Directive 86/609/EEC on the protection of animals used in experimentation.
Carcinogenic induction and administration of NSAIDs
Colon tumors were induced with DMH, with eighteen weekly subcutaneous injections, at a weekly dose of 25 mg/kg of weight. NSAIDs were administered at a daily dose of 3 mg/kg rofecoxib or 30 mg/kg AAS. These drugs were given for 18 weeks postoperatively, starting at 8th postoperative day.
Follow-up and sacrifice
Animals were examined weekly, special attention was paid to their weight, abdominal perimeter, presence and quality of stools, and presence of massive rectal bleeding. All animals were sacrificed in week 20, and their colon was examined in search for colon tumors. Sacrifice was by anesthetic overdose, and the entire colon including 1 cm of the terminal ileum were removed and fixed in a 10% formaldehyde solution for histological examination.
Examination of colon tumors
The entire large intestine of each animal was examined to determine three parameters: number of tumors, tumor surface area, and percentage of tumor surface area (percentage of colon area occupied by tumor tissue). Tumor percentage was determined by estimating total colon surface area versus tumor surface area in the colon, which is the most reliable indicator of the amount of tumor tissue, acting each animal as its own control. The histological study of colon samples also established the histological type of tumors, as well as the histological grade, tumor invasion, and lymph node involvement.
Statistical analysis
The study data were analyzed using the SPSS and G-Stat software programs, and the statistical analysis was performed using one-factor ANOVA models and chi-squared tables. A significant difference was considered for p values < 0.05.
RESULTS
The strict control group was made up of 5 animals without experimental manipulation, which were separated from the rest. In this group of animals, maintained in the same environmental and dietary conditions that the experimental animals, the spontaneous prevalence of colonic tumors was nil. Results for the rats included in the other three groups are analyzed subsequently. These rats were distributed in three experimental groups with carcinogenic induction using DMH (n = 10), carcinogenic modulation with AAS (n = 10), and carcinogenic modulation with rofecoxib (n = 10).
Mortality
The overall mortality rate in this study was of 0%. There was no mortality attributable to colonic induced neoplasm or derived from the administration of the carcinogenic agent or NSAIDs.
Alterations in colon transit
No alterations occurred in gastrointestinal transit; a macroscopic study of the colon after sacrifice disclosed no stenosing neoplasm, either totally or partially.
Number of tumors
A total of 57 colon tumors were found in the 30 animals exposed to DMH, with a mean of 1.9 colonic tumors per animal. Of these 57 tumors, 30 were malignant tumors (adenocarcinomas) and 27 were adenomas. The mean number of adenomas per animal was 0.9, whereas the mean number of adenocarcinomas was 1. The distribution of these tumors is shown in table I. Differences in prevalence of malignant and benign tumors between groups were not significant.

Microscopic tumor surface area
The total colon tumor surface area for all animals included in the study was 16.74 cm2. Of this total area, 7.34 cm2 corresponded to benign tumors and the rest, 9.13 cm2, to adenocarcinomas. Mean tumor surface was greater in the group with induced carcinogenesis when compared with both groups treated with NSAIDs, either with AAS or rofecoxib. This difference applied to all type tumors and to both adenomas and adenocarcinomas separately. Difference was significant between the groups with induced carcinogenesis and group treated with rofecoxib (p < 0.05) in the analysis of all neoplasms, but was not significant when benign and malignant tumors were considered separately, and so it was for the group of rats treated with AAS (Fig. 1).

Microscopic tumor percentage
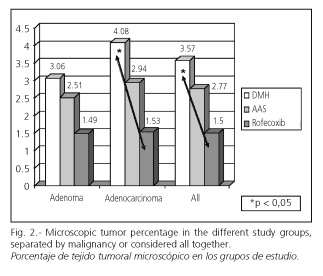
If we consider the NSAIDs group, AAS and rofecoxib, and we analyze all induced tumors, both benign and malignant, NSAIDs produced a significant reduction (p < 0.05) in the microscopic tumor percentage when compared with rats with DMH-induced carcinogenesis. This situation changes when both NSAIDs were considered separately or when benign and malignant tumors were analyzed separately. The average tumor percentage was greater in the group with DMH-induced carcinogenesis versus the other groups. In the group in which rofecoxib was administered to modulate carcinogenesis, a significant reduction in tumor percentage was obtained, which is the most reliable parameter expressing the quantity of tumor induced. This effect was observed for tumors in general but was particularly significant for adenocarcinomas (p = 0,038, LSD, unifactorial ANOVA for mean comparison). However, adenomas did not decrease significantly. Although the group of animals treated with AAS had also a decrease in carcinogenesis, this change was not significant (Fig. 2).
Histologic study
The histological study of the excised colon revealed several different situations: presence of adenocarcinoma, presence of adenoma, colonic nodularity with normal mucosa, and presence of lymphoid hyperplasia in the colonic wall. Lymphoid hyperplasia was found in association with any of the above situations.
Adenomas (n = 27) accounted for 47.37% of induced tumors and were associated with lymphoid hyperplasia in 24 of 27 adenomas (88.89%). Thirty colonic adenocarcinomas were found, 52.63% of induced tumors, of which lymphoid hyperplasia was associated in 20 of these 30 cases (66.66%). Adenocarcinomas were well differentiated in 53.33% of cases, moderately differentiated in 16.67%, and poorly differentiated in 30% of the cases (Table II). As for the invasion extent of adenocarcinomas, most reached the submucosa (53.33%), 30% of them were "in situ", and 16.66% invaded beyond the submucosa (Table III). A total of 120 epicolic nodes (4 per rat) were isolated, 7 of them contained micrometastasis from an adenocarcinoma (5.8%). None of the differences in these parameters was significant between groups.


DISCUSSION
The first evidence of a relationship between NSAIDs and colon cancer was obtained in an epidemiological study conducted by Kune in 1988 (17), where a lower incidence of CRC was reported for patients with inflammatory bowel disease who were chronic users of sulfasalazine.
More than 100 studies have been published that evaluate the use of NSAIDs in the chemoprevention of drug-induced CRC in animal models, and over 85% of them have showed that NSAIDs reduce the prevalence and multiplicity of these tumors (18,19).
In our study, we show that when both NSAIDs, AAS and rofecoxib, and all type tumors, both benign and malignant, are considered a significant reduction (p < 0.05) in the microscopic tumor percentage is seen in comparison with the DMH group. This effect of NSAIDs in the prevention of colic carcinogenesis is well known.
With regard to selective COX-2 inhibitors, the first evidence of their efficacy in animals come from studies conducted in Min (Multiple Intestinal Neoplasia) rats bearing a dominant mutation in the APC (Adenomatous Colon Polyposis) gene, which are characterized by the development of multiple intestinal adenomas at an early age (20). In these animals, the administration of celecoxib caused a greater reduction in the number of adenomas when compared to piroxicam. The administration of rofecoxib also causes similar effects in these rats (20-22). The effects of selective COX-2 inhibitors have also been studied in rats exposed to azoxymethane, a potent colon carcinogen. In these models of drug-induced carcinogenesis, aberrant foci of colon crypts were reduced by 40-49% when celecoxib was administered. Subsequent studies have confirmed significant reductions in the incidence of CRC and in its multiplicity in animals exposed to carcinogens and treated with celecoxib (23,24). Most of these studies have been carried out with celecoxib, but there are few studies with rofecoxib, which has a 2-fold greater selectivity for COX-2 than celecoxib.
The compound MK-0966, rofecoxib, undergoes rapid absorption in rats and attains its peak blood concentration 30 minutes after administration. Dose greater than 5 mg/kg does not increase drug plasma levels in rats (25). It is primarily excreted in the bile, and metabolites derived from oxidation, glucuronoconjugation, or reduction of the drug are not active in terms of inhibition of COX, either 1 or 2 (26). The dosage used in this study, 3 mg/kg of body weight, is high when compared to typical doses used for arthritis and acute pain, but according to clinical studies on its pharmacokinetics, COX-2 inhibition is greater at higher doses, and the tolerability of these high doses was acceptable (27).
Most studies only refer to the number of colon tumors and omit parameters as important as tumor surface area and percentage of tumor area. When not referring to aberrant crypts or foci of displasy, but to induced colon tumors, we believe that the microscopic tumor percentage should be evaluated because it is the only parameter relating the amount of tumor tissue to the size of colon studied. The drug regimen used in our study was 25 mg/kg of DMH for 18 weeks, with sacrifice of animals in week 20, which has been shown to be more effective than other induction regimens using the same drug (28,29) and also to shorten the duration of the study.
In this study, we found that 3 mg/kg rofecoxib significantly reduced the tumor percentage of malignant colon tumors, drug-induced adenocarcinomas, when compared with the DMH-treated control group. This result is consistent with a study recently published by Oshima et al. showing that rofecoxib inhibited polyposis in APCdelta716 mice, a strain of mice that develop intestinal polyposis and secondary carcinomas (22). Although AAS induced a reduction in carcinogenesis when compared with controls, and there were also differences between AAS and rofecoxib, these differences were not significant. In previous studies including a greater number of animals, an inhibitory effect on induced carcinogenesis by non-selective NSAIDs was demonstrated.
Differences between study groups regarding degree of differentiation, invasion extent, and lymph-node involvement were not significant.
As rofecoxib causes a reduction in drug-induced colon carcinogenesis in rats, we believe that this drug should be assessed in future studies of colorectal cancer.
We believe that the dose that should be tested in clinical trials must be greater than those currently used. In two prominent clinical studies currently ongoing -the one by Becerra et al (30) in metastatic CRC and the other by David et al (31) in stage-II and -III CRC, still in its recruitment phase-, the dose of rofecoxib ranges from 25 to 50 mg daily, afar from those employed in this experimental study. The dose of 3 mg/kg of body weight employed in this study represents an approximate dose of 150-200 mg daily for adults between 50 and 70 kg, 3 to 4-fold greater than that currently used in clinical trials. These doses are well tolerated, as shown by initial phase-I and -II pharmacocinetic and security studies (25-27).
ACKNOWLEDGEMENTS
This study has been co-supported by Asociación Española Contra el Cáncer and Junta Balear. We want to thank to the Departamento de Fisiología Animal de la Universitat de les Illes Balears (Prof. R. Rial, Prof. A. Gamundí) for their laboratory and environmental support.
REFERENCES
1. Arnaud JP, Schloegel M, Ollier JC, Adloff M. Colorectal cancer in patients over 80 years of age. Dis Colon Rectum 1991; 34: 896-8. [ Links ]
2. McGregor JR, Galloway DJ, McCulloch P, George WD. Anastomotic suture materials and implantation metastasis: an experimental study. Br J Surg 1989; 76 (4): 331-4. [ Links ]
3. Domínguez F, Riera JR, Junco P, Tojo S, Díaz-Faes M. Influencia en el pronóstico a corto plazo de la sobreexpresión de la proteina p53 en carcinomas colorrectales. Rev Esp Enferm Dig 1994; 86 (5): 796-802. [ Links ]
4. García JC, Cugat E, Angás J, González FJ, Reverter JC, Lacy AM. Cáncer colorrectal: resultados de un protocolo de seguimiento. Cir Esp 1993; 53 (6): 430-3. [ Links ]
5. García JA, Morcillo MA, Vázquez JL, Zaragoza C, Moltó M, Cámara J. Cáncer colorrectal en el anciano. Experiencia de los 5 primeros años en un Servicio de Cirugía de un hospital comarcal. Cir Esp 1996; 60: 256-7. [ Links ]
6. Lieberman DA. Screening for colorectal cancer. Clin Cornerstone 2002; 4: 1-10. [ Links ]
7. Benamouzig R, Chaussade S. La chimioprevéntion du cancer colorectal. Presse Med 2002; 31: 124-8. [ Links ]
8. Sheehan KM, Sheahan K, O'Donoghue DP, MacSweeney F, Conroy RM, Fitzgerald DJ, et al. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA 1999; 282: 1254-7. [ Links ]
9. Vane JR, Bakle YS, Botting RM. Cyclo-oxygenases 1 and 2. Annu Rev Pharmacol Toxicol 1998; 38: 97-120. [ Links ]
10. Brooks P, Emery P, Evans JF, Fenner H, Hawkey CJ, Patrono C, et al. Interpreting the clinical significance of the differential inhibition of cyclo-oxygenase-1 and cyclo-oxygenase-2. Rheumatology 1999; 38: 779-88. [ Links ]
11. Lipsky PE, Brooks P, Crofford LJ, DuBois R, Graham D, Simon LS, et al. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch Intern Med 2000; 160: 913-20. [ Links ]
12. Williams CS, Mann M, DuBois R. The role of cyclo-oxygenases in inflammation, cancer and development. Oncogene 1999; 18: 7908-16. [ Links ]
13. DuBois RN, Radhika A, Reddy BS, Entingh AJ. Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors. Gastroenterology 1996; 110: 1259-2. [ Links ]
14. Bamba H, Ota S, Kato A, Adachi A, Itoyama S, Matsuzaki F. High expression of cyclooxygenase-2 in macrophages of human colonic adenoma. Int J Cancer 1999; 83: 470-5. [ Links ]
15. Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, et al. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res 2000; 60: 293-7. [ Links ]
16. Kargman SL, O'Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res 1995; 55: 556-2559. [ Links ]
17. Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res 1988; 48: 4399-404. [ Links ]
18. Wargovich MJ, Chen CD, Harris C. Inhibition of aberrant crypt growth by nonsteroidal anti-inflammatory agents an differentiation agents in the rat colon. Int J Cancer 1995; 60: 515-9. [ Links ]
19. Rao CV, Rivenson A, Simi B. Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res 1995; 55: 1464-72. [ Links ]
20. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the Apc gene. Science 1992; 256: 668-70. [ Links ]
21. Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res 2000; 60: 5040-4. [ Links ]
22. Oshima M, Murai N, Kargman S, Arguello M, Luk P, Kwong E, et al. Chemoprevention of intestinal polyposis in the Apc ?716 mouse by rofecoxib, a specific cyclooxygenase-2 inhibitor. Cancer Res 2001; 61: 1733-40. [ Links ]
23. Reddy BS, Rao CV, Seibert K. Evaluation of cyclooxygenase-2 inhibitor for potential chemopreventive properties in colon carcinogenesis. Cancer Res 1996; 56: 4566-9. [ Links ]
24. Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res 1998; 58: 409-12. [ Links ]
25. Halpin RA, Geer LA, Zhang KE, Marks TM, Dean DC, Jones AN, et al. The absorption, distribution, metabolism and excretion of rofecoxib, a potent and selective cyclooxygenase-2 inhibitor, in rats and dogs. Drug Metab Dispos 2000; 28: 1244-54. [ Links ]
26. Nicoll-Griffith DA, Yergey JA, Trimble LA, Silva JM, Li C, Chauret N, et al. Synthesis, characterization, and activity of metabolites derived from the cyclooxigenase-2 inhibitor rofecoxib (MK-0966, Vioxx). Bioorg Med Chem Lett 2000; 10: 2683-6. [ Links ]
27. Depre M, Ehrich E, Van Hecken A, De Lepeleire I, Dallob A, Wong P, et al. Pharmacokinetics, COX-2 specificity, and tolerability of supratherapeutic doses of rofecoxib in humans. Eur J Clin Pharmacol 2000; 56: 167-74. [ Links ]
28. Noguera JF, Tortajada C, Morón JM, Plaza A, Amengual I, Pujol JJ. Experimental model for the study of perianastomotic recurrence in colorectal cancer. Rev Esp Enf Dig 2002; 94: 131-4. [ Links ]
29. Noguera JF, Tortajada C, Zurita M, García JC, Álvarez C, Rial R. Nuevo régimen farmacológico para la inducción de tumores cólicos en ratas. Span J Surg Res 2000; 3: 33-5. [ Links ]
30. Becerra CR, Frenkel EP, Ashfaq R, Gaynor RB. Increased toxicity and lack of efficacy of rofecoxib in combination with chemoterapy for treatment of metastatic colorectal cancer: a phase II study. Int J Cancer 2003; 105: 868-72. [ Links ]
31. Kerr DJ. National Institutes of Health. Rofecoxib after surgery in treating patients with stage II or III colorectal cancer. USA (web en línea). Disponible en: http://www.clinicaltrialss.gov (visitada el 4 de enero de 2004). [ Links ]

