










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 n.12 Madrid Dec. 2004
| CLINICOPATHOLOGICAL CONFERENCE |
A young woman with cholestasis and ductopenia
G. Gómez, Y. Rodríguez Gil1, S. Rodríguez Muñoz, P. Sánchez-Pobre, M. Pérez Carreras, F. Colina1 and J. A. Solís Herruzo
Departments of Gastroenterology and 1Pathology. Hospital Universitario 12 de Octubre. Madrid. Spain
Gómez G, Rodríguez Gil Y, Rodríguez Muñoz S, Sánchez-Pobre P, Pérez Carreras M, Colina F, Solís Herruzo JA. A young woman with cholestasis and ductopenia. Rev Esp Enferm Dig 2004; 96: 864-873.
Recibido: 11-11-04.
Aceptado: 11-11-04.
Correspondencia: S. Rodríguez Muñoz. Servicio de Gastroenterología. Hospital Universitario 12 de Octubre. Avda. de Córdoba, s/n. 28041 Madrid. e-mail: sarbeliorm@terra.es
This patient was an 18-year-old woman with a history of psoriasis treated only with topical medication. No other conditions were recorded. She was the daughter of a patient who had undergone liver transplantation for liver cirrhosis secondary to chronic HCV infection and alcohol abuse. The patient presented with no symptoms but reported sporadic pain on her upper right quadrant of the abdomen. Inspection revealed no jaundice, weight loss, anorexia, asthenia, or pruritus. She usually smoked 4-5 cigarettes a day, drank no alcohol, and had no known allergies. She was referred to us after being diagnosed with hypertransaminasemia during routine family follow-up. Serum parameters included: GOT = 105 IU/L; GPT = 97 IU/L; AF = 355 IU/L; GGT = 344 IU/L.
PHYSICAL EXAMINATION
The patient was conscious and oriented, well hydrated and nourished. Her skin and mucous membranes were normal. Her abdomen was soft and undistended with no masses, visceromegalies or tender sites upon palpation.
LABORATORY TESTS
WBC: 6800/mm3 with normal differential count; Hb: 15.0 g/dL; MCV: 88 fl; platelets: 140,000/mm3; coagulation: prothrombine activity = 96%, cephaline = 29 seconds, control = 24 seconds. Blood chemistry: glucemia = 83 mg/dL; LDH = 118 IU/L; GOT = 73 IU/L; GPT = 108 IU/L; alkaline phosphatase: 413 IU/L; GGT = 430 IU/L; bilirubin = 1.05 mg/dL; total protein = 7.7 g/dL; albumin = 5.11 g/dL; creatinine = 0.9 mg/dL; calcium = 9.8 mg/dL; phosphorus = 4.0 mg/dL; uric acid = 5.3 mg/dL; cholesterol = 168 mg/dL; triglycerides = 162 mg/dL; chloride = 99 mg/dL; iron = 104 mcg/dL; ferritin = 111 ng/mL; copper = 76 mcg/dL; ceruloplasmin = 31 mg/dL; Na = 143 mEq/L; K = 5.1 mEq/L. Hepatitis B virus serology: HbsAg, negative; Anti-HBc, positive; anti-HBe and anti-HBs, negative. Hepatitis C virus serology: negative. Amilase: 201 IU/L. Antinuclear, anti-smooth muscle, antimitochondrial, and anti-LKM antibodies: negative. Alphafetoprotein: 7.0 ng/mL; cANCA and pANCA antibodies: negative. 24-hour urine porphyrins: below 200 mcg.
ABDOMINAL ULTRASONOGRAPHY
Liver with normal size, smooth borders, homogeneous parenchyma, and slightly increased echogenicity. Porta vein within normal values, and homogeneous spleen with normal borders. Bile ducts, gallbladder, pancreas, and kidneys were all normal. Conclusion: possible fat-infiltrated liver and/or chronic liver disease.
COMMENTS AND OUTCOME
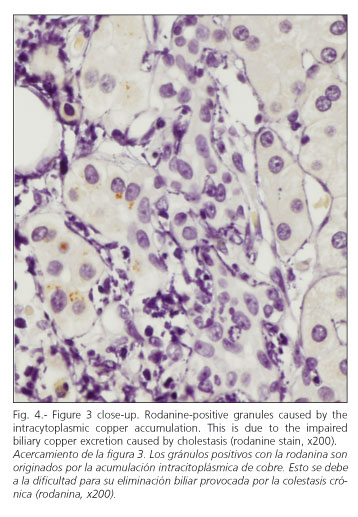
A percutaneous liver biopsy was carried out to complete the patient's examination. It revealed a liver parenchyma with altered architecture, fibrous portal expansion (Fig. 1), and development of mostly incomplete septa with only a few complete porto-portal fibrous bridges. Spatial relationships were adequately preserved between portal tracts and centroacinar veins. No interlobular ducts were seen in over 50% of portal spaces (6 of 11), with presence of hepatic arteries (Fig. 2). Furthermore, portal spaces had a mild mixed inflammatory infiltrate with predominant lymphocytes. The few remaining interlobular ducts had an irregular biliary epithelium. Within the limiting periportal trabecule foci of collagen-entrapped hepatocytes were seen. Periportal hepatocytes were clearly changed and had copper deposits (Figs. 3 and 4). In the remaining lobule, liver cells had their typical characteristics or showed mild hyperplastic features. No positive globules were seen using the periodic acid-Schiff (PAS) stain. No viral inclusions were apparent. Bile pigmentation, steatosis, and siderosis were absent. Endoscopic retrograde cholangiopancreatography, colonoscopy and colon biopsies were uneventful.



Dr. Gonzalo Gómez
In summary, here we have an asymptomatic 18-year-old woman in which a dissociated cholestasis and cytolysis pattern was incidentally found. Laboratory tests and studies searching for chronic liver disease were normal, but ultrasonography showed information suggesting the presence of a chronic liver condition, which was confirmed by histology. We should emphasize the fact that no interlobular ducts were identified in more than 50% of portal spaces.
In our view, the differential diagnosis of this patient should rely on the presence of ductopenia, which is a critical finding.
Congenital causes of ductopenia, particularly Alagille's syndrome, are to be excluded in view of the absence of other characteristics typically associated with this syndrome (1,2), including: facial phenotype with protruding forehead and sunken eyes, defective costal arches, peripherally stenotic or hypoplastic lungs, cardiovascular abnormalities, delayed growth, mental retardation, delayed puberty, voice or bony changes, and renal impairment. The child-onset non-syndromic form of ductopenia has a more aggressive clinical course, with a mortality rate of 50% during the first year of life (3). The case reported here may be consistent with a late-onset non-syndromic form of this disease (4,5).
Regarding alpha1-antitrypsin deficiency, our patient had no history of neonatal or infantile cholestasis (6), as well as no respiratory condition suggesting obstructive pulmonary disease, vasculitis, ulcerative colitis, glomerulonephritis, or skin lesions, which are associated with this deficiency on occasion (7). In addition, electrophoresis revealed no decrease in alpha1-globulins, and histology found no PAS-positive alpha1-antitrypsin inclusions.
Regarding drugs as potential causative agents of this patient's condition (8,9), only topically-administered agents against psoriasis (such as emollients, keratolytic agents, ditranol, corticoids, retinoids, and vitamin D derivatives) were recalled, which makes it difficult for them to be responsible for liver injuries encountered.
Autoimmune hepatitis is ruled out by using the International Autoimmune Hepatitis Group scoring system as revised in 1999 (10), since our patient scored below the 10 points required for diagnosis probability.
Other overlapping autoimmune conditions may be excluded as well because of inadequate scores and lack of antimitochondrial antibodies (AMA) or cholangiographic changes (11).
Another possibility is primary biliary cirrhosis (PBC), particularly in women. Nonetheless, age at the time of diagnosis is usually older, between 50 and 60 years; even though cases have been identified with this age, our patient had no signs or symptoms suggestive of this condition (asthenia, pruritus, hepatomegaly, splenomegaly, hyperpigmentation, jaundice, xanthelasmas). Thus, should our patient have a PBC she would then fall into the category of asymptomatic patients who are usually diagnosed at later ages, which represents 25% (12). We do not see any associated autoimmune conditions either. Alkaline phosphatase is usually higher, and even though histology might suggest a stage III, no granulomas are seen and porto-portal fibrosis is well established altogether. The fact that AMAs are negative does not exclude disease, but a series by Ludwig et al. including 597 patients with primary biliary cirrhosis, of which 35 were negative for AMAs, identified that 96% had positive antinuclear antibodies (ANA) or anti-smooth muscle antibodies (ASMA) (13). Some authors identified this situation (AMA-negative PBC) as corresponding to autoimmune cholangitis (9,14). In a series including 20 patients with autoimmune cholangitis (15), Albert et al. found two subjects with ANA or ASMA negativity. However, when compared to our case report, this group's characteristics had much higher serum AST, alkaline phosphatase, and bilirubin levels. Furthermore, mean age at diagnosis was older. In year 2000, Castellano and Sánchez-Pobre analyzed 203 reports on autoimmune cholangitis (16) and found these very similar to PBC, although negative for AMA and commonly positive for ANA. ASMA positivity was less common, as confirmed by Sánchez-Pobre and Solís (17). These two conditions should also be excluded since ANAs, AMAs and ASMAs are negative, and evidence is not consistent with the aforementioned data.
Small-duct sclerosing cholangitis should also be considered, even in the absence of clear diagnostic criteria. However, in previous series such as that reported by Angulo et al. (18), in which this condition was compared to primary sclerosing cholangitis, no relevant differences in age, gender, clinical manifestations, laboratory findings or histology were encountered, except for a higher percentage of ductopenia and copper deposition in primary sclerosing cholangitis. Patients without inflammatory bowel disease who were diagnosed with small-duct sclerosing cholangitis have been reported, but histological findings already pointed to this condition (19). Our patient was younger than usual at the time of diagnosis, had no manifestations suggestive of inflammatory bowel disease, and both colonoscopy and biopsies showed negative results; also, alkaline phosphatase and GGT were only slightly high, pANCAs were negative, ERCP was normal and pathology revealed no periductal fibrosis, even if this histological change was only seen in 45% of small-duct sclerosing cholangitis cases reported by Angulo et al. (18). All these data, together with an absence of other findings, led us to exclude this condition.
Another cause is Behcet's disease, which is more uncommon but more easily excluded on clinical grounds, since our patient meets none of the diagnostic criteria put forward by the International Group for the Study of Behcet's Disease (20), namely oral ulcers, genital ulcers, skin lesions, ocular lesions, and a positive pathergy reaction.
Regarding sarcoidosis as a secondary cause of ductopenia, this condition usually involves males (21), and our patient had no pulmonary manifestations, chest x-rays were normal, and the liver biopsy showed no granulomas. These have been identified in around 75% of patients with sarcoidosis in a number of series, despite the fact that liver manifestations occur in only 11-12.8% of patients with this disease (22); no asteroid or Schaumann bodies were identified either, and these have been reported also in liver sarcoidosis (23).
Among tumor-related causes histiocytosis X and Hodgkin's disease should be assessed. As Hubscher et al. (24) described, ductopenia may also develop in Hodgkin's disease. On occasion, the patient may even present with liver-related symptoms (25), and thus it should be considered for differential diagnosis. The patient here had no infectious symptoms, which are common in cell-mediated immunodeficiency; no adenopathies or organomegalies were identified upon palpation. Furthermore, ultrasounds revealed no visceromegalies or space-occupying lesions, thus rendering unlikely that intrahepatic cholestases secondary to Hodgkin's disease be found in the absence of anatomic changes to the liver (9); her blood count was normal, with no anemia, leukocytosis, eosinophilia or lymphopenia, and ESR remained unchanged.
As regards histiocytosis X no involvement of the bone, skin, brain with diabetes insipidus, or lung was found to suggest its presence (26); no hemogram changes, hepatomegaly, or splenomegaly were seen on the physical examination or ultrasounds either, ERCP revealed no extrahepatic bile duct involvement, and a liver biopsy showed no erythrophagocytosis.
A thorough history taking allows to rule out CMV-related cholangitis or repeat cholangitis since this patient had no previous symptoms suggesting it, no risk factors for immunosuppression, and a normal blood count. Ischemic hepatitis may also be ruled out in a young, non-smoking, non-hypertensive, non-dyslipemic patient with no manifestations to suggest ischemia at other levels, no previous surgery, and obviously no graft-versus-host disease from liver transplantation or acute rejection following a bone marrow transplant.
Once secondary causes have been excluded, we may safely conclude that our patient meets all inclusion criteria (cholestasis and 50% ductopenia) and no exclusion criteria (absence of granulomas, cholangitis, inflammatory bowel disease, or neoplasm) for the diagnosis of adult idiopathic ductopenia.
DISCUSSION OF HISTOPATHOLOGICAL FINDINGS
Dra. Yolanda Rodríguez Gil
These histopathological findings allow us to infer that this is a chronic liver disease, given the presence of fibrosis (27). She also had stainable copper, which is a sign of chronic cholestasis (28). Bile is the main route for copper excretion, since copper deposits develop in the liver in diseases manifesting with persistent bile stasis. Prolonged cholestasis is required to give rise to copper deposits big enough to be detected by histochemical methods, as demonstrated in previously discussed series of patients with Wilson's disease (29).
Ductopenia directs the diagnosis towards a cause of cholangiopathy
In addition to cholangiopathy, the loss of interlobular ducts is seen in malformations suchs as Alagille' syndrome, chronic liver graft rejection, graft-versus-host disease, sarcoidosis, and Hodgkin's disease. The former three may be safely excluded in our case since none of the multiple malformations accompanying Alagille's syndrome was present, and the patient had received no transplantation. The presence of sarcoidosis may be ruled out on histological grounds since no typical granulomas were seen; also, no evidence of the atypical lymphoid infiltrates characterizing Hodgkin's lymphoma was demonstrated.
Cholangiopathies include typical histologic features that may or may not develop depending on tissue samples. Thus in primary biliary cirrhosis and autoimmune cholangitis, which are histologically identical, florid ductal lesions are characteristic. These include a destructive granulomatous cholangitis of mid-sized ducts. Our patient had no duct-related granulomas, not even the duct-linked granulomas that may be often seen in these conditions.
Conventional primary sclerosing cholangitis has obliterating cholangitis has its characteristic lesion; this is a gradual transformation of interlobular ducts into fibrous cords through progressive "onion-skin" fibrosis stages, which may or may not show up in single biopsy samples due to its segmentary nature. Small-duct sclerosing cholangitis is doubtful in the absence of inflammatory bowel disease, in which case it would be undistinguishable from idiopathic ductopenia. Hepatotoxicity-related cholangiopathy is usually associated with non-chronic inflammatory changes and eosinophilia, and exclusion may be based on previous history. Besides the patient's biochemical context, α1-antitrypsin deficiency may also be histologically ruled out because no PAS-positive diastase-resistant globules were seen on using the periodic acid-Schiff with diastase technique, which accumulate in this condition.
After excluding all other potential causes of intrahepatic duct loss, differential diagnosis is limited to idiopathic ductopenias; in view of the patient's age at onset, this may be a case of favorably-evolving non-syndromic idiopathic ductopenia in infancy, or of idiopathic adulthood ductopenia. Regarding the latter condition, our patient meets both inclusion criteria -ductopenia with an artery/duct ratio below 50% (30)- and both histological (no granulomas, no histiocytosis X, no lymphomas) and clinical (no drug exposure, no idiopathic inflammatory bowel disease, no familial cholestasis, absence of anti-mitochondrial antibodies, no bile duct changes seen on imaging techniques) exclusion criteria. Both types of idiopathic ductopenia, in infancy or in adulthood, are histologically undistinguishable (4). They may be told apart by the presence of associated genetic factors, which usually manifests as involved first-degree relatives in non-syndromic idiopathic ductopenia in infancy.
CLINICAL DISCUSSION AND OUTCOME
Dr. S. Rodríguez Muñoz
Based on liver biopsy the patient was diagnosed with idiopathic adulthood ductopenia (IAD), and treated with 1000 mg/day of ursodeoxycholic acid, which managed to completely remove biochemical changes. Her father's explanted liver was reviewed and showed no changes consistent with adulthood ductopenia; however, a younger sister presented with identical biochemical changes 6 months later, and her biopsy was also consistent with adulthood ductopenia. A study of the remaining first-degree relatives has revealed no other cases thus far.
Until 1988, idiopathic adulthood ductopenia remained hidden in the jumble of cholestatic diseases. The identification, classification, and description of most small-duct conditions brought this enigmatic disease into focus. Ductopenia is currently considered an acquired lesion resulting from a progressive destruction of fully developed bile ducts. However, two evolutive courses exist: a) progressive forms for which liver transplant is the only option within 3 to 11 years (30); and b) benign, non-progressive forms that may or may not manifest with recurrent cholestasis episodes (31,32).
The diagnosis of adulthood ductopenia includes both inclusion and exclusion criteria (33). First, the patient must be an adult or adolescent individual, since ductopenia during childhood is a characteristic of infancy ductopenia. Biochemical evidence for cholestasis is mandatory, and liver biopsy must reveal ductopenia (duct/portal space ratio below 0.5) in the absence of granulomas, cholangitis or neoplasms. Secondly, the condition must be idiopathic, that is, must lack any history of cholestasis during childhood, or of exposure to drugs or toxics. The patient must not have inflammatory bowel disease, Hodgkin's disease, or sarcoidosis. Finally, anti-mitochondrial antibodies must be negative, and both colonoscopy and cholangiography should yield normal results. Gender distribution favors males (1.8/1), and most cases occur between 17 and 27 years of age, although age at onset is variable. Etiology is unknown, but there is evidence suggesting that IAD is a syndrome with multiple causes, and differing profiles regarding both age distribution and prognosis.
Patients with earlier presentations may correspond to late-onset forms of ductopenia of childhood. This late onset has been already described and is strengthened by evidence suggesting genetic factors, since 3 more couples of siblings have been identified with this disease in addition to our patient (34). On the other hand, familial association is also common in other small-duct conditions, as is the case with primary biliary cirrhosis and sclerosing cholangitis.
Another group may correspond to a variant of small-duct primary sclerosing cholangitis lacking obliterative cholangitis, or where liver biopsy revealed no obliterative cholangitis, and also lacking large-duct involvement and any association with inflammatory bowel disease. The latter condition may occur at any age but is most common before 40.
In patients older than this other etiologies are most commonly involved; for instance, a viral cholangitis such as that occasionally induced by hepatitis C virus (35) or other unidentified viruses. This should be considered in the absence of autoimmunity phenomena. Lastly, the presence of non-suppurative cholangitis has been demonstrated both in patients with autoimmune hepatitis (36) and patients with autoimmune cholangitis (37). In such cases the presence of autoantibodies allows to differentiate this condition from adulthood ductopenia. However, patients with autoimmune cholangitis or hepatitis and no autoantibodies have also been described, in whom differentiation from adulthood ductopenia is impossible. The latter patients have a better prognosis, whereas younger patients are more likely to experience a progressive course leading to liver transplantation. Because of her age, family history, and baseline biopsy, our patient may be included amongst those with a poorer prognosis. However, a more favorable outcome may be expected from the complete response of the cholestatic pattern, and her uneventful follow-up with no signs of liver disease progression or portal hypertension for 7 years.
REFERENCES
1. Alagille D, Estrada A, Hadchovel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic displasia): review of 80 cases. J Pediatr 1987 Feb; 110: 195-200. [ Links ]
2. Deutsch GH, Sokol RJ, Stathos TH, Knisely AS. Proliferation to paucity: evolution of bile duct abnormalities in a case of Alagille syndrome. Ped Develop Pathol 2001; 4: 559-63. [ Links ]
3. Alagille D. Management of paucity of interlobular bile ducts. J Hepatol 1985; 1: 561-5. [ Links ]
4. Bruguera M, Llach J, Rodés J. Nonsyndromic paucity of intrahepatic bile ducts in infancy and idiopathic ductopenia in adulthood: the same syndrome? Hepatology 1992; 15: 830-4. [ Links ]
5. Pereda T, Gavilán F, Giráldez A, Sayago M, Serrano J, Gómez MA, et al. Hereditary nonsyndromic paucity of intra hepatic bile ducts as an indication for liver transplantation. Transplantation Proceedings 2003; 35: 719-20. [ Links ]
6. Nemeth A. Pathogenesis of liver disease in alpha1-antitrypsin deficiency. Acta Paediatr Suppl 1994; 5: 393. [ Links ]
7. Cosme A, Ojeda E, Torrado J, Carrera A, Castiella A, Zapata E. Alteraciones hepáticas por déficit de alfa1-antitripsina en adultos. Estudio de 5 pacientes y análisis de los casos publicados en la bibliografía española. Gastroenterol Hepatol 2003; 26: 251-6. [ Links ]
8. Degott C, Feldmann G, Larrey D, et al. Drug-induced prolonged cholestasis in adults. A histological semiquantitative study demonstrating progressive ductopenia. Hepatology 1992; 15: 244-51. [ Links ]
9. Kim WR, Ludwig J, Keith D. Variant forms of cholestasic diseases involving small bile ducts in adults. Am J Gastroenterol 2000; 95: 1130-8. [ Links ]
10. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999; 31: 929-38. [ Links ]
11. Czaja AJ. Frequency and nature of the variant syndromes of autoimmune liver disease. Hepatology 1998; 28: 360-5. [ Links ]
12. Talwalkar JA, Lindor KD. Primary biliary cirrhosis. The Lancet 2003; 362: 53-61. [ Links ]
13. Lacerda MA, Ludwig J, Dickson ER, Jargensen RA, Lindor KD. Antimitochondrial antibody-negative primary biliary cirrhosis. Am J Gastroenterol 1995; 90: 247-9. [ Links ]
14. Salo J, Caballería L, Bruguera M, Pares A, Rodés J. ¿Colangitis autoinmune o cirrosis biliar primaria sin anticuerpos antimitocondriales? Gastroenterología y Hepatología 1992; 20: 5-10. [ Links ]
15. Czaja AJ, Carpenter HA, Santrach PJ, Moore SB. Autoimmune cholangitis within the spectrum of autoimmune liver disease. Hepatology 2000; 31 (6): 1231-8. [ Links ]
16. Castellano G, Sánchez-Pobre P. Colangitis autoinmune. Gastroenterología y Hepatología 2000; 23. Supl 1: 14-9. [ Links ]
17. Sánchez-Pobre P, Solís Herruzo JA. Colangitis autoinmune, localización nosológica. Rev Esp Enferm Dig 2003; 95: 791-4. [ Links ]
18. Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: a long term follow-up study. Hepatology 2002; 35: 1494-500. [ Links ]
19. Broome U, Glaumann H, Lindstom E, Almer S, Prytz H, Sandberg-Gertzén H, et al. Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangities. J Hepatol 2002; 36: 586-9. [ Links ]
20. International study group for Behcet's disease. Lancet 1990; 335: 1078-80. [ Links ]
21. Blich M, Edoute MY. Clinical manifestations of sarcoid liver disease. J Gastroenterol Hepatol 2004; 19: 732-7. [ Links ]
22. Israel HC, Kataria YP. Clinical aspects of sarcoidosis. Sarcoidosis. Orlando (FC): Grune & Satrtton. 1985. p. 7-23. [ Links ]
23. Ishak KG. Sarcoidosis of the liver and bile ducts. Mayo Clin Proc 1998; 73: 467-72. [ Links ]
24. Hubscher SG, Lumley MA, Elias E. Vanishing bile duct syndrome: a possible mechanism for intrahepatic cholestasis in Hodgkin's lymphoma. Hepatology 1993; 17: 70-7. [ Links ]
25. Ripoll C, Carretero L, Sabin P, Álvarez E, Marrupe D, Bañares R. Colestasis idiopática asociada a ductopenia progresiva en dos pacientes con linfoma de Hogkin. Gastroenterol Hepatol 2002; 25: 313-5. [ Links ]
26. Arceci RJ, Longley BJ, Emanuel PD. Atypical cellular disorders. Hematology 2002: 297-314. [ Links ]
27. Sherlock S. Predicting progression of acute type-B hepatitis to chronicity. Lancet 1976; 2: 354-6. [ Links ]
28. García-Ligero R, García Molinero MJ, Colina Ruizdelgado F. Incidencia de los depósitos de cobre en hepatopatías colestásicas. Su valor diagnóstico en relación con la cirrosis biliar primaria. Morfología Normal y Patológica. Sec B 1981; 5: 247. [ Links ]
29. Blasco A, Domínguez P, Colina F, Castellano G. Enfermedad de Wilson. Revisión histológica de 7 pacientes y valor de la positividad del cobre hístico en relación con otras hepatopatías. Med Clin (Barc) 1992; 98: 207-21. [ Links ]
30. Ludwig J, Weisner RH, La Russo NF. Idiopathic adulthood dutopenia. A case of chronic cholestasis liver disease and biliary cirrhosis. J Hepatol 1988; 7: 193-9. [ Links ]
31. Faa G, Van Eyken P, Demelia L, Vallebona E, Costa V, Desmet VJ. Idiopathic adulthood ductopenia presenting with chic recurrent cholestasis. A case report. J Hepatol 1991; 12: 14-20. [ Links ]
32. Moreno A, Carreño VV, Cano A, González C. Idiopathic biliary ductopenia in adults without symptoms of liver disease. N Eng J Med 1997; 336: 835-8. [ Links ]
33. Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc 1998; 73: 285-91. [ Links ]
34. Hartake J, Horle A, Ishli N, Okuno F. Familial intrahepatic cholestatic cirrhosis in adults. Gastroenterology 1985; 89: 202-9. [ Links ]
35. Leftcowitch JH, Schiff ER, Davis GL, Perrilo RP, Lindsay K, Bodenhelmer HC, et al. Pathological diagnosis of chronic hepatitis C: a multicenter comparative study with chronic hepatitis B. Gastroenterology 1993; 104: 595-603. [ Links ]
36. Okuno T, Seto Y, Takino T. Chronic active hepatitis with histological features of primary biliary cirrhosis. Dig Dis Sci 1987; 32: 775-9. [ Links ]
37. Goodman ZD, McNalty PR, Davis DR, Ishak KG. Autoimmune cholangitis: a variant of primary biliary; clinicopathological and serologic correlations in 200 cases. Dig Dis Sci 1995; 40: 1232-42. [ Links ]

