










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.98 no.8 Madrid ago. 2006
POINT OF VIEW
Hepatitis C and insulin resistance: steatosis, fibrosis and non-response
Hepatitis C y resistencia a la insulina: esteatosis, fibrosis y no respuesta
M. Romero-Gómez
Unit for the Clinical Management of Digestive Diseases.
Hospital Universitario de Valme. Seville, Spain
ABSTRACT
Insulin resistance is more often seen in hepatitis C than in other liver diseases, including non-alcoholic steatohepatitis. The Homeostasis Model for Assessment [HOMA= fasting insulin (mUI/ml) * fasting glucose (mmol/L) / 22.5] has proved useful in the measurement of insulin sensitivity in euglycemic patients. Cross-sectional and case-cohort studies support a role for hepatitis C as a factor implied in the development of type-2 diabetes in high-risk patients (male patients, older than 40 years, and overweight). In transgenic mice models the HCV core protein has been found to induce insulin resistance via TNF production. Insulin resistance has been associated with steatosis development and fibrosis progression in a genotype-dependent manner. In genotype-1 patients, the mechanisms by which insulin resistance promotes fibrosis progression include: a) steatosis; b) hyperleptinemia; c) increased TNF production; and d) impaired expression of PPARγ receptors. Indeed, insulin resistance has been found as a common denominator to the majority of features associated with difficult-to-treat patients. Patients with cirrhosis, obesity, coinfected with HIV, and Afro-American, all of them showed insulin resistance. Insulin resistance strongly influences sustained response rates, at least in genotype-1 patients. Insulin resistance decreases during and after treatment in patients that achieved virus C clearance. Moreover, the incidence of type-2 diabetes seems to be lower in responders than in non-responders. In summary, hepatitis C promotes insulin resistance and insulin resistance induces steatosis, fibrosis, and interferon resistance. The treatment of insulin resistance by decreasing hyperinsulinemia could improve sustained response rates in patients with chronic hepatitis C treated with peginterferon plus ribavirin.
Key words: Hepatitis C. Insulin resistance. Steatosis. Fibrosis.
Introduction
Insulin resistance is a major feature of the metabolic syndrome. Resistance emerges as a consequence of the inability of insulin to induce its effect on glucose metabolism. Thus, an abnormally large amount of insulin is required to achieve a biological response. The hyperinsulinemic state induces several abnormalities in the liver, endothelium, and kidneys, and is the main feature in the metabolic syndrome. Currently, some evidence supports a relationship between insulin resistance and hepatitis C. Indeed, insulin resistance is more often seen in patients with chronic hepatitis C than in weight-matched healthy controls, and correlates with fibrosis progression. Besides, virus C infection may induce insulin resistance by blocking intracellular signaling, and lastly, insulin resistance has been associated with a decreased rate of sustained response to peginterferon alpha plus ribavirin in patients with chronic hepatitis C.
Insulin sensitivity
Insulin, after binding its receptor, induces the phosphorylation of receptor substrates in the liver and muscles, and induces several steps toward the transactivation of GLUT-4, which increases glucose uptake by cells and its storage as glycogen, and inhibits the net production of glucose by the liver, thus blocking glycogenolysis and neoglycogenesis. Moreover, insulin promotes lipid storage by inhibiting lipolysis. When insulin is unable to induce glucose uptake, pancreatic beta-cells increase insulin production and the hyperinsulinemic state prevents hyperglycemia. Thus, insulin resistance depends on insulin secretion and insulin sensitivity. The relationship between both events is not linear but hyperbolic. Insulin secretion increases when insulin sensitivity decreases. At a certain level, increasing insulin secretion does no longer improve insulin sensitivity. Thus, insulin concentrations are usually high early in type-2 diabetes, but have also been seen in many subjects who are not obese or diabetic, but who have other metabolic syndrome components such as hypertriglyceridemia or arterial hypertension (1).
Methods for the measurement of insulin resistance
The best method to measure insulin sensitivity is the hyperinsulinemic euglycemic clamp. This method measures the quantity of glucose required to maintain normoglycemic levels during insulin infusion. Briefly, a perfusion of 60 mU/kg/min of insulin is started, and glucose levels are measured every 5 minutes. To avoid hypoglycemia, glucose (20%) was infused to maintain levels between 90 and 100 mg/dl. Whole-body glucose uptake (M value) depends on the glucose infused during the last 30 minutes of the test. Lower M values (lower glucose requirements) are associated with insulin resistance. However, the clamp method is expensive, labor-intensive, and uncomfortable for practical use in clinical medicine.
A simple mathematical model named Homeostasis Model for Assessment (HOMA = fasting insulin (mUI/ml) * fasting glucose (mmol/L)/22.5) has proved useful in the measurement of insulin sensitivity in euglycemic patients. However, this measurement is not useful once blood glucose levels begin to increase, as it lacks accuracy. Normal plasma insulin concentrations are not standardized in healthy controls. Thus, HOMA has not been standardized, and normality ranges in healthy people remain unexplored. In previous reports a HOMA < 2 has been considered "completely" normal, and higher than 4 as a pre-diabetic state. However these thresholds are arbitrary and require further consensus.
Mechanisms of insulin resistance in hepatits C
Several factors have been involved in the development of the insulin resistance syndrome. Hepatic and muscle insulin resistance have been described. Fat storage in the muscles may induce insulin resistance, as free fatty acids have been found to promote the serine-residue phosphorylation of insulin substrate receptor-1 in myocytes, thus inhibiting phosphatidyl inositol-3 kinase (PI3K), and to block the translocation of GLUT-4, thus preventing glucose uptake by these cells. In the liver, the serine phosphorylation of insulin receptor substrate-2 inhibits Akt phosphorylation and the activity of glycogen synthase kinase-3, thus preventing glycogen synthesis and promoting glucose release.
Islet adaptation by pancreatic beta cells is a compensating mechanism to maintaining glucose levels, and is responsible for hyperinsulinemia. Several factors have been proposed as mediators in islet adaptation: glucose, free fatty acids, autonomic nervous system, or glucagon-like peptide 1. Although hyperglycemia may rise insulin secretion, in patients with insulin resistance hyperinsulinemia persists in spite of normal blood glucose levels. Lastly, when pancreatic islet beta cells cannot secrete insulin enough to maintain glucose homeostasis, type-2 diabetes mellitus develops.
The mechanisms by which hepatitis C induces an increased risk for the development of diabetes are not fully understood. Liver fibrosis has been long considered responsible for the development of insulin resistance and type-2 diabetes in patients with chronic liver disease. Hyperinsulinemia in liver cirrhosis has been reported to be due to a diminished liver insulin extraction as a result of liver dysfunction and not pancreatic hypersecretion. C-peptide and insulin are secreted in equimolar quantities. More than 50% of insulin is degraded in the liver at first pass, whereas C-peptide is degraded in the kidneys (2). Simultaneous measurements of C-peptide and insulin revealed that both insulin resistance and insulin secretion contribute to glucose intolerance in patients with chronic hepatitis C (3). Although insulin resistance correlated with liver fibrosis, insulin resistance was found to be higher in patients with chronic hepatitis C and mild fibrosis (F0-F1) than in healthy controls matched for age, sex, visceral obesity, and body mass index (4). Besides, these differences have been found using simple methods to measure insulin resistance, such as HOMA, or more sophisticated methods like the euglycemic-hyperinsulinemic clamp (5). Moreover, insulin resistance has been found to be higher in patients with chronic hepatitis C than in patients with non-alcoholic steatohepatitis (6). Insulin resistance is considered a key factor in the pathophysiology of NASH. Thus, hepatitis C virus induces insulin resistance. In a transgenic animal model using mice expressing the core HCV protein, insulin resistance developed, whereas it did not appear in non-transgenic mice. Also, a fat-rich diet induces type-2 diabetes in transgenic but not wild animals. Hepatitis C virus may induce an overproduction of TNF, responsible for the phosphorylation of serine residues at insulin-receptor substrates 1 and 2, and an enhanced production of the suppressor of cytokines (SOC3). SOC inhibits the phosphorylation of Akt and phosphatidyl inositol-3 kinase (PI3K). All these impairments in the intracellular signaling of insulin may block the transactivation of GLUT-4, preventing the uptake of glucose by cells. The role of HCV core protein, TNF, and SOC production in the development of insulin resistance has been demonstrated in animal transgenic models. In fact, in transgenic mice expressing the HCV core protein, insulin resistance appears to result from an overproduction of TNF. However, in transgenic animal models unable to express SOC-3, in spite of core HCV protein expression, insulin resistance does not develop. Indeed, in transgenic mice, TNF correlates with hyperinsulinemic states, and the blockade of TNF production by anti-TNF drugs such as infliximab also prevents insulin resistance. Therefore, the mechanism inducing insulin resistance by hepatitis C virus includes: TNF production, serine phosphorylation of IRS, and overexpression of SOCs. Furthermore, an overproduction of TNF in patients with chronic hepatitis C has usually been found (15), and correlated with higher fibrosis progression and impaired antiviral response to interferon alpha. TNF is a proinflammatory and profibrogenic cytokine, and the risk for the development of insulin resistance and then of DMT2 is greater in patients with higher TNF production. Indeed, TNF levels correlated with fasting insulin levels and insulin resistance indexes such as HOMA, independently of liver fibrosis stage (7).
Type 2 diabetes mellitus and hepatitis C
The development of type-2 diabetes mellitus depends on environmental, genetic and diet-related factors. Type-2 diabetes mellitus is more often seen in patients with chronic liver diseases than in the general population (8). Data supporting an association between diabetes mellitus and hepatitis C include: a) cross-sectional studies that found an increased prevalence of diabetes mellitus in patients with chronic hepatitis C, or a higher prevalence of hepatitis C in patients with diabetes mellitus; and b) case-cohort studies to analyze the development of diabetes mellitus during follow-up in patients with hepatitis C against non-infected patients, including follow-up after orthotopic liver transplantation. In a cross-sectional survey including 9,841 persons, Mehta et al. found that HCV-positive persons who were older than 40 years had an increased risk for type-2 diabetes mellitus, more than 3-fold when compared to persons without HCV infection. However, no difference was seen between HBV infected subjects and the general population (9). Diabetes mellitus has been more often seen in cirrhotic patients (10). However, in a cohort of 45 non-cirrhotic patients with chronic hepatitis C the prevalence of type 2 diabetes mellitus was 33%, higher than in the matched control group and in a group of patients with chronic hepatitis B (11). Besides, in a large retrospective study diabetes mellitus was present in 23.6% of patients with hepatitis C, and in 9.4% of those with hepatitis B (12). On the other hand, anti-HCV antibodies have more often been detected in patients with diabetes mellitus than in the general population, although the prevalence of antibodies varies from 3 to 6-9% (13). However, some confounding factors, including economic status, race, and body mass index, have not always been included in these analyses. Some data support the hypothesis that in patients with a high risk for the development of type-2 diabetes (male patients, older than 40 years, and overweight) HCV infection plays a role in the development of diabetes mellitus during follow-up. In a cohort of 1,084 patients followed-up for 9 years 548 cases developed diabetes mellitus. The presence of hepatitis C was associated with a greater development of diabetes, but solely in high-risk diabetes patients (14). Besides, in a cohort of 2,327 cases, hepatitis C infection increased three times the rate of diabetes mellitus development during follow-up in patients aged between 35 and 49 years (15). Thus, HCV infection may promote type-2 diabetes in high-risk populations.
Recently, in a Spanish study 525 chronic hepatitis C patients treated with peginterferon plus ribavirin were followed up after treatment, and the incidence of impaired fasting glucose and development of type-2 diabetes was greater in non-responders than in sustained responders even after a multivariate analysis including confounding variables such as previous type-2 diabetes in relatives, age above 40 years, and male sex. Thus, the hypothesis that hepatitis C virus clearance induces a decrease in the insulin resistance index during short-term follow-up was confirmed by studying the incidence of type-2 diabetes during long-term follow-up (16).
Steatosis, fibrosis and insulin resistance
Hepatocyte-related steatosis is a common feature found in the liver of patients with chronic hepatitis C. The mechanisms involved in the development of steatosis seem to be genotype-dependent. In patients infected by genotype 3, steatosis emerges as a cytopathic effect of the virus itself, while with genotype 1 steatosis seems to be an expression of metabolic syndrome. In genotype 3, steatosis degree correlates with liver HCV quantification and serum viral load. In genotype 1, steatosis depends on leptin levels and insulin resistance. Recently, insulin resistance has been found to be involved in the development of steatosis in 331 non-diabetic genotype-1 patients. Insulin resistance together with gender and gammaglutamyl-transpeptidase (a surrogate marker of TNF levels) was independently associated with moderate/severe steatosis in this cohort (17).
Insulin resistance and hyperinsulinemia have been found in association with liver fibrosis in hepatitis C. Indeed, high serum glucose levels have been found associated with an increased rate of fibrosis progression, even greater than overweight (18). The mean insulin resistance index increases with stage of fibrosis (19) and may help differentiate fibrosis stages. Recently, Sud et al. (20) proposed an index to predict fibrosis containing age, cholesterol, gammaglutamyl-transpeptidase, and alcohol consumption together with insulin resistance. This index improves a little the diagnosis accuracy of other tests, including routine biochemical parameters such as platelets or transaminases, but the accuracy was lower than 90% and only half of patients could be correctly classified as near one half showed intermediate scores. The mechanisms through which insulin resistance promotes fibrosis progression include: 1. Steatosis. 2. Hyperleptinemia. 3. Increased TNF production; and 4. Impaired expression of PPARγ receptors. 1. Hepatocyte-related steatosis induces fibrosis progression: in a cohort of patients with chronic hepatitis C, the presence of steatosis in the first biopsy was associated with a higher fibrosis progression rate, irrespective of genotypes (21). Besides, steatosis is more often seen in patients with advanced fibrosis (22). 2. Hyperleptinemia has been usually found in patients with insulin resistance, also hepatic stellate cells showed specific leptin receptors, thus leptin may play a role in the activation of HSC and fibrosis progression (23,24). 3. TNF production is enhanced in hepatitis C and has been implicated in the development of insulin resistance; also, TNF levels were related to fibrosis progression, owing to their ability to activate HSC and promote collagen deposits. Moreover, TNF may inhibit PPAR activity (25); and 4. In patients with hepatitis C an impaired expression of PPARγ receptors has been found (26). PPAR agonists inhibit inflammation and fibrosis progression by blocking the activation of the redox-sensitive transcription factor NFkB and TGFß1 (27).
Insulin resistance in HIV-HCV coinfection
In HIV-infected patients insulin resistance depends on host-, virus- and drug-related factors. Adverse metabolic effects have been found in all antiretroviral drug classes. Protease inhibitors (PI) and nucleoside reverse transcriptase inhibitor (NRTI) induce insulin resistance during treatment (28). Increased insulin resistance has been found in HIV+/HCV+ coinfected patients. Factors implied in the development of insulin resistance in HIV-infected patients include: HCV infection, PI- and/or NRTI-based therapy, age, HIV and cytokine dysregulation as induced by chronic infection, and genetic predisposition (29). Moreover, the development of type-2 diabetes mellitus was twice as high in coinfected patients versus those infected with HIV but not with HCV (30). Duong et al. (31) reported a higher HOMA in coinfected patients than in HIV-infected cases without hepatitis C. However, in a large study no differences were seen between coinfected and not coinfected patients (30). In a recent study including 127 coinfected patients who underwent antiviral therapy (peginterferon plus ribavirin) and 85 hepatitis C subjects, the insulin resistance index was higher in coinfected patients in spite of a lower body mass index and lower baseline glucose levels. Besides, in a genotype-dependent manner, insulin resistance was strongly associated with steatosis development and impaired sustained response rate in genotype-1 coinfected patients. No association between insulin resistance and steatosis or sustained response was seen in genotype-2 or -3 coinfected patients (32).
Lastly, a history of diabetes in relatives, an increased body mass index, and HCV-infection have been found as independent variables associated with diabetes development in HIV patients, supporting the hypothesis that HCV infection promotes diabetes in high-risk patients.
Insulin resistance and response to peginterferon plus ribavirin
In healthy volunteers, insulin resistance could be detected after the first injection of interferon alpha (33). Besides, in patients with chronic hepatitis C interferon alpha induces insulin resistance in the first two weeks, mainly owing to a decrease in hepatic glucose uptake (34). This effect has been related to the proinflammatory repertoire of cytokines induced by interferon. However it is a transitory effect because insulin resistance was not found at month three of therapy (35) or the end of therapy (36). In patients with chronic hepatitis C receiving peginterferon plus ribavirin insulin resistance measured by HOMA decreased in patients with HCV RNA clearance at month 6, but not in non-responders. At the end of follow-up sustained responders showed a significantly lower HOMA in comparison to their baseline insulin resistance index. However, in relapsers the HOMA index increased, and their levels at the end of follow-up were not different from those at baseline. These data support a connection between HCV replication and insulin resistance, and HOMA decreased when the virus was eradicated. In addition, the incidence of diabetes type 2 is different in cured patients than in non-responders, supporting a better control of insulin resistance after hepatitis C virus clearance (16).
Genotype and viral load have been found as the most important viral factors influencing the response to peginterferon plus ribavirin. Host factors include genes such as HLA, weight, body mass index, hepatocyte steatosis, age, Afro-American ethnicity, and fibrosis.
Several genes and polymorphisms such as HLA-B44 have been found to be related to the possibility of curing the disease (37). On the other hand, features of metabolic syndrome such as overweight, steatosis, and fibrosis have been reported as independently associated with response. The influence of steatosis in the chance of curing hepatitis C has been controversial for a long time. Steatosis impairs SVR rate in patients with genotype 1, but not in those with genotype 3. Thus, the possibility of finding steatosis as an independent variable depends on the balance between genotypes in the cohort. In fact, in a multivariate analysis including steatosis together with fibrosis, body mass index, genotype and insulin resistance, neither steatosis nor body mass index were found independently associated with response, but insulin resistance, genotype and fibrosis were. In genotype 1, the sustained response rate was 32% in patients with insulin resistance (HOMA > 2), versus 60% in patients with a HOMA lower than 2 (38). However, in a recent cohort including 331 non-diabetic genotype-1 patients insulin resistance was not found to be associated with response. Besides, in 52 patients from the UK also treated with peginterferon plus ribavirin, the HOMA index was significantly higher in non-responders than in patients with sustained response (39). Thus, insulin resistance emerges as the most important host factor in the prediction of response in non-diabetic patients treated with the best available option -peginterferon plus ribavirin. Interestingly, insulin resistance has been found a common denominator to the majority of features associated with difficult-to-treat patients. Patients with cirrhosis, obesity, HIV coinfection and Afro-American showed all insulin resistance.
Hyperinsulinemia induces interferon resistance
Peginterferons induce their antiviral activity via extracellular receptor binding. The interferon alpha signaling pathway involves the activation of Janus kinase (Jak1) and tyrosine kinase (Tyk2), initiated by the binding of peginterferon alpha-2 to the interferon heterodimeric receptor complex (IFNAR1/IFNAR2), which leads to the activation of their downstream substrates, signal transducers and activators of transcription (STAT 1 and STAT2). Activated STAT then assembles as a multimeric complex and translocates into the nucleus, where it binds interferon alpha-2-stimulated response elements in the promoters of interferon alpha-2-stimulated genes (40). Recently, in a replicon model using Huh-7 cells transfected with full-length HCV-RNA, interferon alpha blocked HCV replication. However, when insulin (128 µU/ml, similar to that seen in the hyperinsulinemic state in patients with metabolic syndrome) was added to interferon, the ability to block HCV replication disappeared, and PKR synthesis was abolished (41). Thus, hyperinsulinemia induces interferon resistance. In this experiment, a blockage of PI3K by LY294002 avoided the interference of insulin, supporting that insulin-induced interferon resistance is mediated by PI3K (Fig. 1).
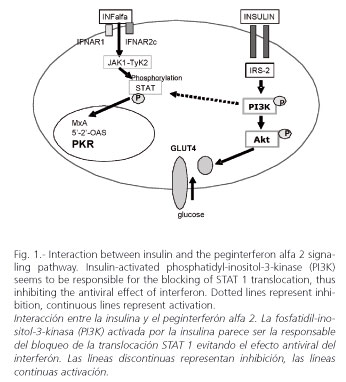
In summary, hepatitis C promotes insulin resistance and insulin resistance induces interferon resistance, steatosis and fibrosis progression in a genotype-dependent manner. With genotype 1, insulin resistance decreases sustained response rates and increases the risk for steatosis and fibrosis progression in both coinfected HCV+/HIV+ and hepatitis C patients. However, the impact of insulin resistance for genotypes other than 1 seems to be weak. The treatment of insulin resistance -decreasing hyperinsulinemia- may improve sustained response in genotype-1 patients with chronic hepatitis C when treated with peginterferon plus ribavirin.
Acknowledgements
The author thanks Dres. J. Salmerón (Hospital San Cecilio, Granada), R. Andrade (Hospital Virgen de la Victoria, Málaga), C. Fernández-Rodríguez (Hospital Fundación Alcorcón, Madrid), M. Diago (Hospital General de Valencia, Valencia), C. Tural (Hospital Germans Trías i Pujol, Badalona), M. C. Martínez-Sierra (Hospital Puerta del Mar, Cádiz), R. Sola (Hospital del Mar, Barcelona), R. Planas (Hospital Germans Trías i Pujol, Badalona), B. Clotet (Hospital Germans Trías i Pujol, Badalona) for the works performed together on insulin resistance in hepatitis C in HCV-HIV-coinfected patients. We would also like to thank Drs. F. Recio, J. Castillo, M. Cruz, I. Camacho, and M. M. Viloria (Hospital Universitario de Valme, Sevilla) for their kindly measuring of insulin resistance.
References
1. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365: 1415-28. [ Links ]
2. Kruszynska YT, Home PD, McIntyre N. Relationship between insulin sensitivity, insulin secretion and glucose tolerance in cirrhosis. Hepatology 1991; 14: 103-11. [ Links ]
3. Narita R, Abe S, Kihara Y, Akiyama T, Tabaru A, Otsuki M. Insulin resistance and insulin secretion in chronic hepatitis C virus infection. J Hepatol 2004; 41: 132-8. [ Links ]
4. Hui JM, Sud A, Farrell GC, et al. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression. Gastroenterology 2003; 125: 1695-704. [ Links ]
5. Yazicioglu G, Isitan F, Altunbas H, et al. Insulin resistance in chronic hepatitis C. Int J Clin Pract 2004; 58: 1020-2. [ Links ]
6. Kawaguchi T, Yoshida T, Harada M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol 2004; 165: 1499-508. [ Links ]
7. Maeno T, Okumura A, Ishikawa T, et al. Mechanisms of increased insulin resistance in non-cirrhotic patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2003; 18: 1358-63. [ Links ]
8. Zein CO, Levy C, Basu A, Zein NN. Chronic hepatitis C and type II diabetes mellitus: a prospective cross-sectional study. Am J Gastroenterol 2005; 100: 48-55. [ Links ]
9. Mehta SH, Brancati FL, Sulkowski MS, Strathdee SA, Szklo M, Thomas DL. Prevalence of type 2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Ann Intern Med 2000; 133: 592-9. [ Links ]
10. Thuluvath PJ, John PR. Association between hepatitis C, diabetes mellitus, and race. a case-control study. Am J Gastroenterol 2003; 98: 438-41. [ Links ]
11. Knobler H, Schihmanter R, Zifroni A, Fenakel G, Schattner A. Increased risk of type 2 diabetes in noncirrhotic patients with chronic hepatitis C virus infection. Mayo Clin Proc 2000; 75: 355-9. [ Links ]
12. Caronia S, Taylor K, Pagliaro L, et al. Further evidence for an association between non-insulin-dependent diabetes mellitus and chronic hepatitis C virus infection. Hepatology 1999; 30: 1059-63. [ Links ]
13. Simo R, Hernández C, Genesca J, Jardi R, Mesa J. High prevalence of hepatitis C virus infection in diabetic patients. Diabetes Care 1996; 19: 998-1000. [ Links ]
14. Mehta SH, Brancati FL, Strathdee SA, et al. Hepatitis C virus infection and incident type 2 diabetes. Hepatology 2003; 38: 50-6. [ Links ]
15. Wang CS, Wang ST, Yao WJ, Chang TT, Chou P. Community-based study of hepatitis C virus infection and type 2 diabetes: an association affected by age and hepatitis severity status. Am J Epidemiol 2003; 158: 1154-60. [ Links ]
16. Romero Gómez M, Fernández-Rodríguez C, Diago M, et al. La curación de la hepatitis C reduce el riesgo de desarrollo de hiperglucemia y diabetes mellitus tipo 2. Gastroenterol Hepatol 2006; (en prensa). [ Links ]
17. Cammà C, Bruno S, Di Marco V, et al. Insulin resistance is associated with steatosis in non-diabetic patients with genotype 1 chronic hepatitis C. Hepatology 2006; 43: 64-71. [ Links ]
18. Ratziu V, Munteanu M, Charlotte F, Bonyhay L, Poynard T; LIDO Study Group. Fibrogenic impact of high serum glucose in chronic hepatitis C. J Hepatol 2003; 39: 1049-55. [ Links ]
19. Fartoux L, Poujol-Robert A, Guechot J, Wendum D, Poupon R, Serfaty L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut 2005; 54: 1003-8. [ Links ]
20. Sud A, Hui JM, Farrell GC, et al. Improved prediction of fibrosis in chronic hepatitis C using measures of insulin resistance in a probability index. Hepatology 2004; 39: 1239-47. [ Links ]
21. Ratziu V, Trabut JB, Poynard T. Fat, diabetes, and liver injury in chronic hepatitis C. Curr Gastroenterol Rep 2004; 6: 22-9. [ Links ]
22. Romero-Gómez M, Castellano-Megias VM, Grande L, et al. Serum leptin levels correlate with hepatic steatosis in chronic hepatitis C. Am J Gastroenterol 2003; 98: 1135-41. [ Links ]
23. Otte C, Otte JM, Strodthoff D, et al. Expression of leptin and leptin receptor during the development of liver fibrosis and cirrhosis. Exp Clin Endocrinol Diabetes 2004; 112: 10-7. [ Links ]
24. Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in dipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol 2005; 166: 1655-69. [ Links ]
25. Sung CK, She H, Xiong S, Tsukamoto H. Tumor necrosis factor-alpha inhibits peroxisome proliferator-activated receptor gamma activity at a posttranslational level in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 2004; 286: G722-9. [ Links ]
26. Dharancy S, Malapel M, Perlemuter G, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology 2005; 128: 334-42. [ Links ]
27. Burgess HA, Daugherty LE, Thatcher TH, et al. PPAR gamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol 2005; 288: L1146-53. [ Links ]
28. Taiwo BO. Insulin resistance, HIV infection, and anti-HIV therapies. AIDS Read 2005; 15: 171-6, 179-80 (review). [ Links ]
29. Shikuma CM, Day LJ, Gerschenson M. Insulin resistance in the HIV-infected population: the potential role of mitochondrial dysfunction. Curr Drug Targets Infect Disord 2005; 5: 255-62. [ Links ]
30. Visnegarwala F, Chen L, Raghavan S, Tedaldi E. Prevalence of diabetes mellitus and dyslipidemia among antiretroviral naïve patients co-infected with hepatitis C virus (HCV) and HIV-1 compared to patients without co-infection. J Infect 2005; 50: 331-7. [ Links ]
31. Duong M, Petit JM, Piroth L, et al. Association between insulin resistance and hepatitis C virus chronic infection in HIV-hepatitis C virus-coinfected patients undergoing antiretroviral therapy. J Acquir Immune Defic Syndr 2001; 27: 245-50. [ Links ]
32. Romero Gómez M, Solà R, Tural C, et al. La resistencia a la insulina disminuye la respuesta sostenida a interferón pegilado más ribavirina en pacientes coinfectados VIH/VHC. Gastroenterol Hepatol 2006 (en prensa). [ Links ]
33. Koivisto VA, Pelkonen R, Cantell K. Effect of interferon on glucose tolerance and insulin sensitivity. Diabetes 1989; 38: 641-7. [ Links ]
34. Imano E, Kanda T, Ishigami Y, et al. Interferon induces insulin resistance in patients with chronic active hepatitis C. J Hepatol 1998; 28: 189-93. [ Links ]
35. Ito Y, Takeda N, Ishimori M, Akai A, Miura K, Yasuda K. Effects of long-term interferon-alpha treatment on glucose tolerance in patients with chronic hepatitis C. J Hepatol 1999; 31: 215-20. [ Links ]
36. Tai TY, Lu JY, Chen CL, et al. Interferon-alpha reduces insulin resistance and beta-cell secretion in responders among patients with chronic hepatitis B and C. J Endocrinol 2003; 178: 457-65. [ Links ]
37. Romero-Gómez M, González-Escribano MF, Torres B, et al. HLA class I B44 is associated with sustained response to interferon + ribavirin therapy in patients with chronic hepatitis C. Am J Gastroenterol 2003; 98: 1621-6. [ Links ]
38. Romero-Gómez M, Viloria MM, Andrade RJ, et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology 2005; 128: 636-41. [ Links ]
39. D'Souza R, Sabin CA, Foster GR. Insulin resistance plays a significant role in liver fibrosis in chronic hepatitis C and in the response to antiviral therapy. Am J Gastroenterol 2005; 100: 1509-15. [ Links ]
40. Grace MJ, Cutler D. Pegylating IFNs at His-34 improves the in vitro anti-viral activity through the JAK/STAT pathway. Antiviral Chem Chem 2004; 15: 287-97. [ Links ]
41. Sanyal AJ, Chand N, Comar K, Mirshahi F. Hyperinsulinemia block the inhibition of HCV replication by interferon: A potential mechanism for failure of interferon therapy in subjects with HCV and NASH. Hepatology 2004; 40: 179A. [ Links ]
![]() Correspondence:
Correspondence:
Manuel Romero-Gómez.
Unidad de Gestión Clínica de Enfermedades Digestivas.
Hospital Universitario de Valme.
Ctra. de Cádiz, s/n. 41014. Sevilla.
Fax: 955 015 899.
E-mail: mromerog@supercable.es
Recibido: 17-01-06
Aceptado: 25-01-06

