










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.99 no.8 Madrid Ago. 2007
High grade primary adrenal intravascular large B-cell lymphoma manifesting as Addison disease
Linfoma intravascular de alto grado de células B grandes y origen suprarrenal que se manifiesta en forma de enfermedad de Addison
J. Venizelos, D. Tamiolakis1, M. Lambropoulou1, G. Alexiadis2, G. Petrakis1 and N. Papadopoulos1
Department of Pathology. Ippocration General Hospital of Salonica. 1Department of Histology-Embryology.
Democritus University of Thrace. 2Diagnostic Radiology Center of Alexandroupolis. Thrace, Greece
ABSTRACT
We report a rare case of a 68 aged male who presented with adrenal failure and was diagnosed of high grade large B-cell lymphoma primarily arising in the adrenal glands. The patient was administrated with additional chemotherapy but he passed away 7 months later due to infection in the lungs. Intravascular lymphoma should be suspected in patients with bilateral adrenal masses who present with rapidly progressive adrenal insufficiency.
Key words: B-cell lymphoma. Addison disease. Adrenal glands.
RESUMEN
Publicamos el caso poco frecuente de un varón de 68 años de edad que debutó con insuficiencia adrenal y fue diagnosticado de linfoma de alto grado de células B grandes ubicado principalmente en las glándulas suprarrenales. Al paciente le administraron quimioterapia adicional, pero falleció 7 meses después de infección pulmonar. El linfoma intravascular debe sospecharse en los pacientes con masas suprarrenales bilaterales que presenten insuficiencia adrenal rápidamente progresiva.
Palabras clave: Linfoma de células B. Enfermedad de Addison. Glándulas adrenales.
Introduction
It is known that, development of adrenal failure requires destruction of more than 90% of the adrenal gland tissue (1). Most cases of adrenal insufficiency caused by malignant neoplasms are due to metastases from a lung, breast, or stomach primary, and malignant melanoma (2). Malignant lymphoma could occur in any body organ. However, non- Hodgkin lymphoma (NHL) arising in the endocrine system represents only 3% of extranodal malignant lymphomas, and is usually confined to the thyroid gland (3). Adrenal infiltration at the time of presentation is rare and difficult to quantify. Surprisingly secondary involvement by lymphoma of the adrenal glands in autopsy findings rises 35% (4). Primary adrenal lymphoma tends to be predominant in males compared to females (2.5-7 times), occurs with an average age of 61 years (38-81), morphology of large cell lymphoma and a B cell phenotype with a small number of T cell phenotype (5). Of the 70 cases reported so far, bilateral cases constitute 70% of the total adrenal lymphoma (5). Symptoms may occur due to the tumor itself or adrenal failure.
Case study
A 68-year-old male was diagnosed 7 months ago with anaemia due to duodenal ulcer and subsequently treated with anti-ulcer drugs and ferrum, but he failed to respond. Four months prior to admission, he complained of nausea and developed fever and diarrhoea. He presented with generalized pigmentation, on admission. Clinical examination showed organomegaly without peripheral lymphadenopathy. Because of anaemia, lymphocytosis, and hepatosplenomegaly, the patient underwent an abdominal CT investigation that revealed bilateral adrenal masses, hepatic and splenic enlargement, but no other pathology. Laboratory tests showed normal complete blood count and no abnormal results on urinalysis. Biochemical tests demonstrated a high LDH level (427IU/l). Serum electrolyte analysis revealed no hyponatremia, but elevated values of serum potassium and serum CRP. Endocrinological data showed plasma ACTH 820 pg/ml, plasma cortisol 4.4 μg/day, and urinary-cortisol 2.3 μg/day. Furthermore, urinary 17 KS, urinary 17-OHCS, serum DHEA-S, serum estradiol, and plasma aldosterone were low, confirming adrenal failure.
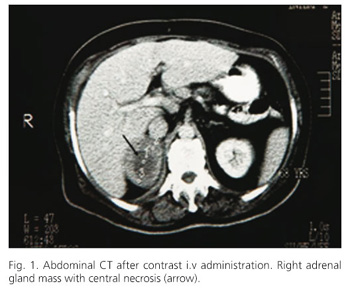
Abdominal CT detected two masses measuring 5 cm and 2 cm in the right and left adrenal gland respectively, with comparable and inhomogeneous density after contrast administration. The right adrenal revealed hypodense center due to necrosis (Fig. 1). No celiac lymph node enlargement was shown. Magnetic resonance imaging revealed low signal intensity on T1 weighted images and high signal intensity with heterogeneous appearance in both of the adrenal glands on T2-weighted images. We suspected metastatic tumors and searched the entire body for the primary lesion with negative results. 67Ga scintigraphy did not reveal any abnormal uptake in the adrenals or other organs.
The patient was administrated with hydrocortisone replacement therapy resulting in immediate improvement of symptoms. A surgical excision of the left adrenal gland followed.
Pathological findings
Gross examination showed a 9.5 x 7 x 1.5 oval, encapsulated, rubbery, brown-white coloured tumor. No adrenal tissue was recognised. Histopathologically (Fig. 2), there was complete and diffuse infiltration by large pleomorphic cells with round hypochromatic nuclei, 2 to 3 prominent nucleoli at the periphery of the nuclear envelope and a slightly eosinophylic cytoplasm. There were also medium sized cells some of them with nuclear grooves, a few immunoblasts, and plasma-like cells. Mitotic figures were frequent.

Immunohistopathology
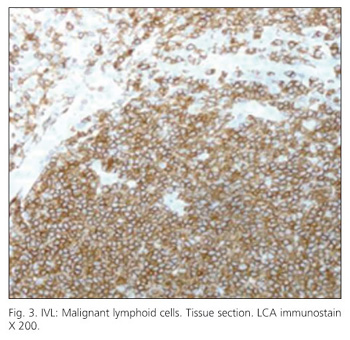
Immunohistochemical staining demonstrated that most tumor cells were positive for LCA (leukocyte common antigen) (Fig. 3), B-cell markers (CD20, CD79a), but although stained for bcl-2, they did not stain for CD5. Malignant cells were surrounded by endothelial (CD34 positive) cells (Fig. 4).

Based on pathological assessment, the final diagnosis was intravascular large B-cell lymphoma, a subtype of extranodal diffuse large B-cell lymphoma characterized by the presence of lymphoma cells in the lumina of small vessels.
Following the resection of the left adrenal gland, serum electrolytes improved under the replacement therapy with hydrocortisone (30 mg/day). The pigmentation gradually disappeared, and plasma ACTH decreased to 18.7 pg/ml. However, after the operation, persistence of symptoms of general fatigue, appetite loss, low-grade fever, steal sweat and elevation of CPR and LDH was observed. The activity of NHL remained high. Subsequent abdominal enhanced CT revealed multiple low intensity areas in the liver and celiac lymph node enlargement, which had not been detected by then. The disease was staged as IVB according to the Ann Arbor system and the patient underwent chemotherapy with CHOP (Cyclophosphamide,doxorubicin, vincristine, and prednisolone), two cycles twice a week, resulting in remission. Seven months later the patient passed away due to infection in the lungs.
Discussion
We treated a 68 year old man who was diagnosed with bilateral primary adrenal malignant lymphoma. The most common manifestations of malignant lymphoma are the involvement of the amygdale, stomach and pharynx, in addition to the lymph nodes, although the involvement of various other organs has been described. Involvement of the adrenal gland in malignant lymphoma cases is reported to be 25% at autopsy (6), but malignant lymphoma arising in the endocrine glands represents only 3% of extranodal malignant lymphomas, and is usually confined to the thyroid gland. Therefore, primary adrenal malignant lymphoma is rare.
The present patient showed typical symptoms and endocrinological findings of adrenal failure. Primary adrenal malignant lymphoma was described in 24 patients of 67 reported cases complicated with adrenal failure including subclinical or partial adrenal failure (7,8). These reports showed certain characteristic features for primary adrenal malignant lymphoma, such as male predilection, involvement of both adrenal glands and being of the diffuse large B cell type. CT and MRI revealed heterogeneous tumors in our case. The heterogeneous tumors on imaging studies are usually tumors with bleeding and necrosis, making it very difficult to differentiate malignant lymphoma from adrenal metastatic tumors or adrenal cortical carcinomas (9,10). Necrosis is associated with rapid tumor development. Surgery was performed to allow rapid histopathological examination under operation for the final diagnosis. The final diagnosis was intravascular lymphoma (IVL) based on histopathological examination. IVL was classified as diffuse large B-cell lymphoma subtype, according to the new WHO classification (11). Several cases of IVL have been reported since the first patient was reported by Pfleger and Tappeiner (12). Generally, antemortem diagnosis of IVL is very difficult because of the exceedingly rapid clinical course and protean presentation of the disease. Additionally, the poor prognosis of IVL results partially from delays in diagnosis. IVL is characterized by massive proliferation of atypical mononuclear cells within arterioles, capillaries, and venules. This disease is therefore rarely diagnosed clinically but could present with various clinical features, such as fever, skin eruptions, neurological symptoms, and weight loss (13,14). Moreover, the most common manifestations of IVL are infiltration of the skin and central nervous system, although infiltration of atypical cells has been described in various organs (15). Numao et al. (16) indicated that primary and secondary adrenal gland infiltration of IVL was detected in 67% of patients diagnosed with IVL.
The sensitivity of IVL to systemic chemotherapy is very high and particularly when early diagnosis is made followed by treatment at an early stage. Therefore, early diagnosis and treatment of IVL is crucial for prognosis. Recent studies showed that de novo CD5+ diffuse large B-cell lymphoma (CD5+ DLBCL) is phenotypically and genotypically different from CD5- DLBCL (17). The survival of patients with CD5+ DLBCL is significantly worse than that of patients with CD5- DLBCL. Moreover, several patients with CD5+ IVL have been described (18,19). However, in the present case the tumor cells did not stain for CD5.
IVL should be suspected in patients with bilateral adrenal tumors who present with rapidly progressive adrenal failure.
References
1. Symmerrs W. Systemic pathology. 2nd ed. London: Churchill Livingstone; 1978. [ Links ]
2. Mor F, Lahav M, Kipper E, Wysenbeek AJ. Addison's disease due to metastases to the adrenal glands. Postgrad Med 1985; J 61: 637-9.
3. Freeman C, Berg JW, Culter SJ. Occurrence and prognosis of extranodal lymphomas. Cancer 1972; 29: 252-60.
4. Straus DJ, Filippa DA, Lieberman PH. The non Hodgkin lymphomas. A retrospective clinical and pathological analysis of 499 cases diagnosed between 1958-1969. Cancer 1983; 51: 101-9.
5. Sachico IW, Tohru A, Nayoyoshi M. Bilateral adrenal lymphoma with hypercalcemia: An autopsy case report with a review of the literature. J Clin Exp Hematopathol 2003; 43 (1).
6. Rosenberg SA, Diamond HD, Jaslowitz B, Craver LF. Lymphosarcoma: A review of 1269 cases. Medicine 1961; 40: 31-84.
7. Isaji M, Kuroda H, Shibata T, et al. A case of adrenal failure due to bilateral adrenal non-Hodgkin lymphoma. Folia Endocrinol Jap 2000; 76 (Supl.): 105-8.
8. Miyabayashi H, Kitano K, Terashima M, et al. B cell malignant lymphoma complicated with partial Addison's disease. Report of a case. Rinsho Ketsueki 1993; 34: 1016-21.
9. Gondoh M, Sakamoto H, Fujii A, et al. A case of primary non-Hodgkin lymphoma of left adrenal gland. Rinsho Hoshasen 1987; 32: 155-8.
10. Hirota H, Takai K, Kohira T, et al. Two cases of primary adrenal malignant lymphoma. Medical 1992; 24: 1483-5.
11. Harris NL, Jaffe ES, Diebold J, et al. The World Health Organization classification of hematological malignancies report of the clinical advisory committee meeting, Airlie House, Virginia November 1997. Mod Pathol 2000; 13: 193-207.
12. Pfleger L, Tappeiner J. On the recognition of systematized endotheliomatosis of the cutaneous blood vessels (reticuloendotheliosis?). Hautarzt 1959; 10: 359-63.
13. Oshiomi K. Neoplastic angioendotheliosis. Igakunoayumi 1992; 162: 115-7.
14. Shiota M, Mori S. Malignant histiocytosis and angioendothelial lymphoma. Naika 1991; 68: 316-9.
15. Mizusawa H, Okaneya T, Yoneyama T, Taniguchi I. Primary malignant lymphoma of the adrenal gland: A case report. Acta Urol Jpn 1995; 41: 991-4.
16. Numao H, Ido K, Narita T, et al. A primary adrenal gland lymphoma that presented as intravascular lymphomatosis. J Cancer Clin 1991; 37: 1821-5.
17. Yamaguchi M, Seto M, Okamoto M, et al. De novo CD5+ diffuse large B-cell lymphoma: A clinicopathologic study of 109 patients. Blood 2002; 99: 815-21.
18. Kanda M, Suzumiya J, Ohshima K, Tamura K, Kikuchi M. Intravascular large cell lymphoma: Clinicopathological, immunohistochemical, and molecular genetic studies. Leuk Lymphoma 1999: 34: 569-80.
19. Yegappan S, Coupland R, Arber DA, et al. Angiotropic lymphoma: an immunophenotypically and clinically heterogeneous lymphoma. Mod Pathol 2001; 14: 1147-56.
 Correspondence:
Correspondence:
Nicolas Papadopoulos
Assoc. Professor in Histology-Embryology
Democritus University of Thrace
Dragana, 68100 Alexandroupolis, Greece.
E-mail: npapad@med.duth.gr
Recibido: 20-03-07.
Aceptado: 28-03-07.

