










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.100 n.8 Madrid Aug. 2008
Pancreatitis crónica autoinmune
Chronic autoimmune pancreatitis
L. Aparisi Quereda
Servicio de Aparato Digestivo. Hospital Clínico de Valencia
Dirección para correspondencia
Introducción
En 1961 Sarles y cols. describen por primera vez, en un grupo de diez pacientes no alcohólicos, las características de un tipo de pancreatitis diferente a los conocidos. Estos tenían una edad media de 60 años, presentaban escaso o nulo dolor abdominal pero sí frecuentemente colestasis, con aumento de las gammaglobulinas séricas. Histopatológicamente el páncreas mostraba una intensa infiltración inflamatoria y fibrosis. Se denominó a esta afección pancreatitis inflamatoria primaria (1). Aunque diferentes autores habían descrito pancreopatías con parecidos síntomas, incluyendo en España a Oliver Pascual y cols. en 1961 (2), es en el Simposio de Marsella-Roma en 1988 donde se la incluye por primera vez como nueva pancreopatía con el nombre de pancreatitis crónica inflamatoria (3). Durante los siguientes años los estudios sobre esta enfermedad fueron escasos y generalmente sobre casos clínicos aislados.
A partir de la década de 1990 cobra interés su estudio, especialmente en Japón. En 1995 Yoshida y cols., resumiendo las publicaciones efectuadas sobre esta enfermedad, señalan en 10 puntos sus características básicas (Tabla I) y proponen la denominación de pancreatitis autoinmune (4), término con el que con más frecuencia se la conoce en la actualidad. No obstante, no ha sido hasta el año 2003 cuando se llega a aceptar internacionalmente como entidad nosológica, describiendo sus características y proponiendo los primeros criterios diagnósticos (5). Además de pancreatitis autoinmune, esta enfermedad ha recibido otras denominaciones, haciendo énfasis los correspondientes autores en los diferentes aspectos significativos de esta (Tabla II).


Etiología y patogenia
Las causas de la pancreatitis autoinmune no son conocidas y sólo recientemente se acepta la existencia de una base autoinmune sistémica, llegando a considerarla como una enfermedad esclerosante IgG4 dependiente (13), fundamentándose en los siguientes hechos:
-Frecuente asociación con otras afecciones autoinmunes, con el mismo patrón histopatológico: colangitis esclerosante secundaria, sialoadenitis esclerosante, nefritis tubulointersticial, neumonitis intersticial (14).
-Frecuente presencia de autoanticuerpos frente a la anhidrasa carbónica II y IV (15-19) y lactoferrina (20), enzimas que normalmente están presentes en los conductos/células acinares del páncreas, glándulas salivares, vías biliares, riñón, etc., órganos igualmente afectados en esta enfermedad. Recientemente se ha matizado la inicial hipótesis que considera la anhidrasa carbónica II y la lactoferrina como los antígenos diana principales de este proceso autoinmune, al observar clínicamente que la presencia sérica de sus correspondientes anticuerpos posee una limitada sensibilidad y especificidad (15,18,19).
-Reproducción experimental de las alteraciones histopatológicas en animales timectomizados e inmunizados con anhidrasa carbónica II o lactoferrina (21).
-Aumento de IgG4 sérica (22) y de células plasmáticas IgG4 + en los tejidos (14). Aunque su presencia es muy constante, cabe según algunos autores la posibilidad de que esta pueda representar sólo una respuesta secundaria a un no identificado gatillo del proceso inflamatorio (23).
-Aumento de linfocitos T activados CD4 y CD8 tanto circulantes como infiltrando los tejidos, en estrecha asociación con los haplotipos HLA DRB1*0405-HLA DRB1*0401, lo que sugiere un proceso autoinmune (20,24).
Definición
No existe aún una definición de esta enfermedad unánimemente consensuada. De las que recientemente se han propuesto, la de los autores de la Clínica Mayo (25) es la que en mi opinión mejor recoge el estado de los conocimientos actuales: "La pancreatitis autoinmune es una enfermedad sistémica fibroinflamatoria que afecta además del páncreas a otros órganos como los conductos biliares, glándulas salivares, retroperitoneo y ganglios linfáticos. Los órganos afectados presentan una infiltración linfoplasmocitaria con abundantes células IgG4 positivas. El proceso inflamatorio responde al tratamiento con esteroides."
Frecuencia
La mayoría de los estudios efectuados a este respecto son retrospectivos, comprendiendo un número de pacientes limitado y por lo tanto con resultados variables. En la tabla III se indican los resultados de los estudios sobre series médicas o quirúrgicas.

Se ha calculado que en las series médicas la prevalencia de pancreatitis autoinmune sobre la pancreatitis crónica fluctúa entre el 4 y el 8%. En España se ha efectuado un estudio multicéntrico sobre 54 pacientes con pancreatitis crónica inicialmente diagnosticadas de idiopáticas y donde finalmente se ha llegado al diagnóstico de pancreatitis autoinmune en 8 casos, lo que constituye el 15% de los pacientes (19). Recientemente Ghazale y cols. han publicado los resultados de un estudio prospectivo de 487 pacientes con sospecha genérica de pancreopatía, en los que se incluía a pacientes con ictericia obstructiva, masas quísticas o sólidas del páncreas, pancreatitis y dolor abdominal. El número de casos con un diagnóstico definitivo de pancreatitis autoinmune fue de 22 pacientes, resultando pues un 4,5% de los sujetos estudiados (29).
Entre las publicaciones sobre pacientes sometidos a cirugía, hay que señalar el estudio de Yadav en 2003, sobre 245 pacientes con pancreatitis crónicas, de los que 45 presentaban una pancreatitis "tumefactiva" (masa pancreática y/o ictericia obstructiva). De estos fueron finalmente diagnosticados como pancreatitis autoinmune 27 casos, lo que representó el 10,6% de todas las pancreatitis crónicas y el 60% de las pancreatitis tumefactivas (11). En un estudio multicéntrico europeo sobre 200 casos con resección de páncreas por pancreatitis crónica, se ha constatado que el 26% de los pacientes tenía una pancreatitis autoinmune (31).
Todos estos estudios vienen a confirmar que es una enfermedad poco frecuente, aunque no tanto como para no tenerla en cuenta a la hora de efectuar un diagnóstico definitivo en una pancreopatía de etiología incierta o con manifestaciones clínicas atípicas.
Características
Las manifestaciones de esta afección pueden agruparse en manifestaciones clínicas, biológicas, morfológicas de imagen e histopatológicas y de respuesta a la terapéutica (Tabla IV).

Manifestaciones clínicas
Uno de los aspectos comúnmente señalado por todos los autores es la escasa frecuencia de dolor, manifestación muy frecuente en la mayoría del resto de pancreopatías, lo que, unido a su más frecuente aparición en la sexta década de edad, ictericia obstructiva y un cuadro constitucional, obligan frecuentemente a considerar como primera hipótesis diagnóstica a una neoplasia de páncreas (32) (Tabla V). Aunque los episodios agudos de pancreatitis son muy infrecuentes, estos han sido descritos en sujetos más jóvenes (13).

El sexo masculino es mucho más frecuente que el femenino en todas las series publicadas. Una de las características frecuentemente reseñada es la asociación a otras afecciones autoinmunes en el 60 al 80% de los casos (Tabla VI). Recientemente se ha calculado que la afectación de las vías biliares extrapancreáticas se da en el 74% de los casos (en forma de colangitis esclerosante secundaria), las glándulas lacrimales o salivares en el 39% (en forma de Sjögren secundario o sialoadenitis), fibrosis retroperitoneal en el 12-15%, artritis reumatoide en el 23%, linfoadenopatías hiliares mediastínicas en el 80%, alteraciones tiroideas en el 22% y neuropatías en el 12% (13,33). Con frecuencia, la aparición de estas alteraciones extrapancreáticas se da de manera metacrónica con el inicio de las pancreáticas (33).

Han sido descritas alteraciones exocrinas y endocrinas en la mayoría de los pacientes (34-37). En las fases iniciales la diabetes es generalmente no insulina-dependiente y reversible tras el tratamiento con corticoides (36), pasando a insulina-dependiente e irreversible con la evolución en los casos no tratados. Igualmente la esteatorrea es constante en las fases avanzadas de la enfermedad (37).
Manifestaciones biológicas
Las manifestaciones biológicas observadas en la pancreatitis autoinmune, según han ido siendo descritas en la literatura, son el aumento de las gammaglobulinas séricas, la presencia de autoanticuerpos y finalmente el aumento de la inmunoglobulina IgG, especialmente la fracción 4 de esta, la IgG4 (Tabla VII).

El aumento de gammaglobulinas en estos pacientes fue la primera alteración biológica señalada en 1961 por Sarles y cols. (1). Posteriormente en la siguiente década y especialmente tras la caracterización clínica de la enfermedad efectuada en 1995 por Yoshida y cols. (4), se concreta entre estas el aumento de las IgG séricas, aunque con escasa especificidad. Finalmente Hamano y cols. en 2001 constatan por primera vez que sólo la fracción 4 de las IgG séricas estaba elevada en la pancreatitis autoinmune, al contrario que en otras afecciones, donde puede plantearse un problema de diagnóstico diferencial con el cáncer de páncreas (22). A partir de los hallazgos de Hamano y cols., el aumento de la IgG4 sérica ha sido considerado como el principal marcador biológico de la pancreatitis autoinmune; no obstante, el aumento de IgG4 no es totalmente específico de la pancreatitis autoinmune, ya que se ha encontrado elevada también en la dermatitis atópica, pénfigo, asma, parasitosis como la filariosis, etc.
En los últimos años se ha analizado por diferentes autores el valor diagnóstico del aumento de la IgG4 sérica en la práctica clínica (19,29,33,38,39), calculándose que su sensibilidad diagnóstica fluctúa entre el 76 y el 92% y su especificidad entre el 95 y el 97% (28,32,39), para unos valores séricos por encima de 135 ó 140 mg/dl (29,40), concluyendo que los valores elevados de esta son característicos de la pancreatitis autoinmune, pero los aumentos moderados pueden darse en otras afecciones como en el cáncer de páncreas. Así se ha observado que cuando las elevaciones de la IgG4 sérica son leves (< 2 veces el límite normal), se obtienen resultados falsamente positivos entre el 5 y 10%, especialmente con el cáncer de páncreas (Tabla VIII) (29) y que, por el contrario, al elevar el punto de corte, obviamente se reduce su sensibilidad diagnóstica a la vez que aumenta la especificidad, 53 y 99% respectivamente, con un valor predictivo positivo del 75% (29). Recientemente Kamisawa y cols. describen la presencia de aumentos de IgG4 sérica e infiltración de plasmacélulas en algún paciente con cáncer de páncreas, por lo que recomiendan que en los sujetos con masa pancreática los valores moderadamente elevados de IgG4 sean interpretados con prevención (13).

Entre el 30 y el 50% de los pacientes presentan valores séricos elevados de Ca 19-9, posiblemente debido a la colestasis (aunque es infrecuente que rebasen tres veces el límite normal) (32,41,42), lo que contribuye a que frecuentemente se plantee un problema de diagnóstico diferencial con el cáncer de páncreas.
La presencia de autoanticuerpos en esta enfermedad ha sido constatada en la descripción de casos aislados desde la década de 1980 y posteriormente en 1995 incluidos en los criterios diagnósticos de Yoshida y cols. (4) (Tabla I). Su valor diagnóstico aislado en la actualidad es escaso y sólo en el contexto del resto de manifestaciones (Tabla VII). Inicialmente Nishimori y cols. en 1994 evidencian en estos enfermos la frecuente presencia de antoanticuerpos séricos y la asociación a otras afecciones autoinmunes (síndrome de Sjögren, sialoadenitis, colangitis esclerosante, etc.), comprobando que dichos autoanticuerpos lo son frente a un antígeno expresado en los conductos de las glándulas exocrinas y especialmente frente al páncreas, proponiendo el concepto de exocrinopatía autoinmune como mecanismo autoinmune en su patogénesis (16). En 1995 el mismo equipo demuestra experimentalmente que este antígeno era la anhidrasa carbónica II (43). En 1996 se comprueba la presencia sérica de anticuerpos antianhidrasa carbónica II en algunos de los pacientes con pancreatitis crónica considerada idiopática hasta ese momento (18). En aquel momento este equipo investigador considera la hipótesis de que estas enfermedades constituyan la manifestación, en diferentes órganos, de una reacción autoinmune frente a un antígeno común expresado en las células de los conductos de los órganos exocrinos y que la anhidrasa carbónica II era el candidato a ser este antígeno diana (18). En los siguientes años fueron publicados varios estudios clínicos donde se analizaba el valor diagnóstico de la presencia de estos autoanticuerpos, llegándose a la conclusión de su escasa especificidad (15,18,19), habiéndose encontrado en otras afecciones como las pancreatitis alcohólicas, síndrome de Sjögren primario, lupus eritematoso, endometriosis, artritis reumatoide, hepatitis viral, etc., por otro lado se constató su ausencia en casos con pancreatitis autoinmune comprobada, calculándose que estos anticuerpos están presentes en sólo el 50% de los casos (20,26,41). Ello, unido al hecho de que la IgG4 sérica se encuentra aumentada en esta enfermedad de manera más sensible y específica (Fig. 1) (Tabla VIII) y a las dificultades técnicas de su determinación, ha hecho que actualmente su aplicación clínica se haya abandonado.
La presencia de autoanticuerpos frente a la lactoferrina (20), así como antinucleares, antimúsculo liso, antimitocondriales o factor reumatoide, se ha descrito en porcentajes diferentes en estos pacientes, de manera no específica (Tabla VII).
Adicionalmente se ha observado un aumento sérico de las transaminasas, enzimas pancreáticas y bilirrubina en las crisis agudas de la enfermedad (20), secundarios a las alteraciones anteriormente descritas.
Alteraciones morfológicas de imagen
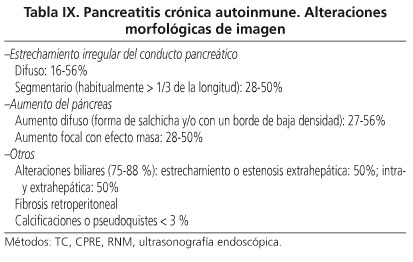
Las alteraciones morfológicas de imagen consideradas básicas en la pancreatitis autoinmune son el aumento difuso del páncreas y el estrechamiento irregular del conducto principal, formando parte de los criterios diagnósticos cardinales aceptados por las principales escuelas (25,39,40) (Tabla IX). Estas alteraciones se corresponden con las alteraciones histopatológicas descritas más adelante.
Parénquima pancreático
Mediante TAC, antes del tratamiento con corticoides, puede observarse un aumento difuso, homogéneo, hipodenso del páncreas, con pérdida del carácter lobulado, semejante a una salchicha cuando está todo el páncreas afectado (en el 26 al 56% de los casos) (25,39,44) (Fig. 2B), pudiendo presentar característicamente un borde liso de baja densidad (Fig. 3I). En otros casos, en el 28 al 50%, el aumento puede ser focal, especialmente en la cabeza del páncreas, en forma de una masa con baja atenuación, denominada por algunos autores pancreatitis tumefactiva (25,32,44,45) (Fig. 2A), lo que, unido a la frecuente aparición del aumento del tamaño de los ganglios regionales, hace que se plantee habitualmente un problema de diagnóstico diferencial con el cáncer de páncreas (11). Es infrecuente observar calcificaciones pancreáticas o pseudoquistes (25,32,44,45).


La utilización de la ecografía endoscópica ha cobrado un creciente interés debido a su capacidad de explorar detalladamente el páncreas y poder efectuar biopsias. Las imágenes mostradas permiten observar también un páncreas "hinchado" con ecoestructura alterada difusamente o localmente (especialmente en la cabeza, como masa focal) y aumento de los ganglios peripancreáticos en el 40% de los casos (> 1 cm). Estos resultados no son totalmente coincidentes con la TAC y complementan a veces los hallazgos de esta (44). El valor diagnóstico de la ecografía transabdominal es limitado dada su poca especificidad, observándose habitualmente un páncreas aumentado hipoecoico (44). No está claramente establecido el valor de la RNM con colangiopancreatografía (44,46); no obstante se ha constatado también un aumento difuso o localizado del páncreas con baja densidad en T1 y alta en T2 (Fig. 4A).

Conductos pancreáticos
Desde las primeras descripciones se ha señalado que en esta enfermedad existe un estrechamiento irregular del conducto principal (4). Este puede ser difuso, a lo largo de todo el conducto principal (Fig. 3III), o localizado y en este caso típicamente el estrechamiento es mayor de un tercio de toda la longitud del conducto, al contrario de lo que habitualmente ocurre en el cáncer de páncreas (Fig. 5) (5,39,40). Con la progresión de la enfermedad, sin tratamiento, este estrechamiento tiende a extenderse a toda la extensión del conducto (46). También ha sido observado que distalmente al estrechamiento raramente existe una dilatación significativa del conducto, a diferencia de lo que comúnmente existe en la pancreatitis crónica etílica o el cáncer de páncreas (25,32,44,45). Estas alteraciones ductales se han observado especialmente tras la colangiopancreatografía endoscópica (5,41,42), siendo más dificultosa la objetivación de estas en las colangiopancreatografías tras RNM (38,39), especialmente a la hora de diferenciar los estrechamientos ductales propios de esta enfermedad de los de otras pancreatopatías (47). La visualización del conducto pancreático puede hacerse más evidente tras la administración de secretina (Fig. 4B, C).

Extrapancreáticamente se han evidenciado alteraciones de imagen en las vías biliares, retroperitoneales, pulmonares, renales y hepáticas en consonancia con su condición de enfermedad multifocal (33,48). Las alteraciones biliares se han observado entre el 75 y el 88% de los casos (33,49), como estrechamientos o estenosis de las vías intra- y extrahepáticas con dilatación proximal, constituyendo una colangitis esclerosante secundaria. Recientemente han sido descritas diferencias colangiográficas con respecto a la colangitis esclerosante primaria, como la mayor frecuencia de estenosis por segmentos, ausencia de apariencia de árbol podado, la dilatación tras los estrechamientos y mayor afectación en el tercio distal del conducto biliar (50,51); adicionalmente sólo en la colangitis de la pancreatitis autoinmune se obtiene una buena respuesta tras el tratamiento con corticosteroides.
Como una manifestación más de la fibrosis multifocal propia de la enfermedad ha sido descrita la presencia de una fibrosis retroperitoneal en un 12% de los casos (Fig. 6) (33), en forma de masa con densidad de tejidos blandos periaórtica, confundiéndose a veces con un proceso maligno (linfoma, sarcoma, carcinomas de otros órganos retroperitoneales), pudiendo afectar a los uréteres y provocar hidronefrosis (52), así como fibrosis en el hilio hepático asociado a hipertensión portal con varices esofágicas (37).

También han sido descritas alteraciones morfológicas y funcionales correspondientes a otras lesiones extrapancreáticas asociadas como sialoadenitis, síndrome de Sjögren secundario, linfoadenopatías hiliares pulmonares, así como imágenes pseudotumorales hepáticas o pulmonares (13,33).
Alteraciones morfológicas histopatológicas
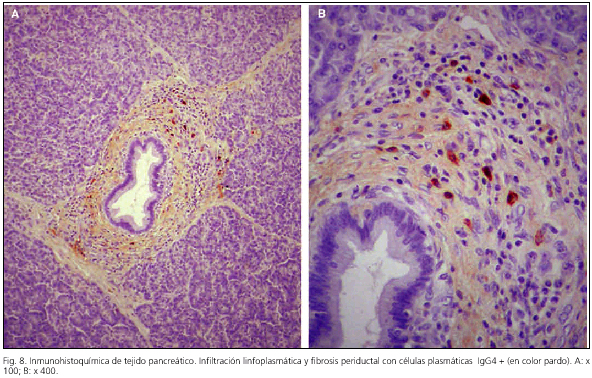
En la actualidad se consideran como alteraciones histopatológicas pancreáticas básicas de la pancreatitis autoinmune una intensa infiltración inflamatoria con una distribución predominantemente periductal, una fibrosis densa difusa (storiform fibrosis) y una flebitis obliterativa (Tabla X) (25,31,39,40). La infiltración está compuesta principalmente de linfocitos y células plasmáticas y la fibrosis es usualmente densa y difusa. La infiltración y la fibrosis se dan tanto en áreas interlobulares como intralobulares, con afectación periacinar, asociadas a atrofia acinar y encapsulamiento de los islotes en estadios avanzados. Los conductos pequeños se afectan generalmente en los estadios avanzados de la enfermedad y las células acinares van siendo progresivamente reemplazadas por la infiltración inflamatoria. La fibrosis con la evolución de la enfermedad da lugar a la atrofia pancreática. Los conductos pancreáticos están estrechados con plegamiento hacia el interior de la luz como consecuencia de la infiltración, la fibrosis y el edema periductal (Fig. 7). En la mitad de los casos de la serie de Zamboni y cols. (31) se observan pequeños núcleos inflamatorios con miofibroblastos entremezclados con haces de colágeno, que en algunos casos ocupan áreas mayores adquiriendo una apariencia pseudotumoral. Ocasionalmente se observan folículos linfoides aislados e infiltrados con eosinófilos. Igualmente se observa característicamente una flebitis obliterativa, afectando a venas de diferente tamaño, con infiltración linfoplasmática y proliferación de fibroblastos alrededor de la pared de la vena (Fig. 7). Zamboni y cols. hacen notar que en los sujetos más jóvenes se da una infiltración granulocítica epitelial con destrucción del epitelio ductal, asociada frecuentemente al padecimiento de una colitis ulcerativa o Crohn (31).


Kamisawa y cols. en 2003 demuestran que en la pancreatitis autoinmune las alteraciones histopatológicas básicas observadas en tejido pancreático se observan también en otros tejidos como en los conductos biliares, ganglios linfáticos, tejido peripancreático y glándulas salivares, y que la infiltración se compone predominantemente de linfocitos T CD4 o CD8 y sobre todo de plasmacélulas IgG4 positivas (48), dando lugar además de la pancreatitis a colangitis esclerosante secundaria, sialoadenitis, fibrosis retroperitoneal, afectación de venas portales o flebitis obliterativa. Este patrón histopatológico ha sido posteriormente observado también en glándulas lacrimales, linfáticos mediastínicos, riñón, hígado, papila duodenal, estomago, intestino, tiroides o articulaciones, dando lugar a síndrome de Sjögren, linfadenopatías hiliares, nefritis, pseudotumor hepático, duodenitis, colitis ulcerosa, tiroiditis, artritis, con una frecuencia variable (13,25,31,33,37,46,53-57).
En los últimos años la demostración de la presencia infiltrativa de plasmocélulas IgG4 ha sido utilizada como prueba diagnóstica confirmatoria de la pancreatitis autoinmune (13,19,25) (Fig. 8). No obstante, también se ha observado que la presencia de plasmacélulas IgG4 + infiltrantes en pequeñas cantidades puede darse también en otras pancreatopatías incluido el cáncer de páncreas, habiéndose calculado que sólo la infiltración intensa (microscópicamente: ≥ 10 plasmacélulas IgG4+/campo), en el contexto de los otros parámetros, supone una gran ayuda en la confirmación diagnóstica (ver más adelante) (58). Recientemente Kamisawa propone el término de "enfermedad esclerosante asociada a IgG4-positiva" para esta entidad nosológica (13).
La obtención de suficiente tejido pancreático como para poder observar estas alteraciones con la histopatología estándar presenta dificultades en la práctica clínica, teniendo en cuenta que en numerosos casos la lesión tiene una distribución focal, limitando el valor diagnóstico de las biopsias con aguja fina ecoendoscópicamente guiadas (13,25,31). Según el grupo de la Clínica Mayo la demostración inmunohistoquímica de una infiltración suficiente de plasmacélulas IgG4 + podría soslayar esta dificultad (25), hecho también cuestionado recientemente (59).
Características según la respuesta a la terapéutica
Desde las primeras descripciones de la pancreatitis autoinmune se señaló que sus manifestaciones clínicas cedían rápida y espectacularmente tras el tratamiento con corticosteroides. Esta característica es muy constante y por ello se ha incluido como prueba diagnóstica en la mayoría de los criterios diagnósticos propuestos en la actualidad (5,23,25,39). Esta respuesta afecta principalmente al componente inflamatorio, siendo la fibrosis frecuentemente permanente.
Especialmente cuando existe ictericia obstructiva o aumento focal del páncreas (masa pancreática), la respuesta se considera como positiva si tras el tratamiento (Tabla XI) se obtiene una clara mejoría de la ictericia y reducción franca del páncreas (25,60).

Las molestias abdominales y la colestasis ceden con rapidez. Las alteraciones pancreáticas de imagen, aumento de tamaño o estrechamiento del conducto pancreático se normalizan en pocas semanas (25,35,39,45), considerándose esta respuesta como específica de la enfermedad (25,39) (Fig. 3). Los valores séricos de IgG4 se normalizan o se reducen significativamente (Fig. 9) (37,45). Igualmente se ha descrito que en las fases iniciales de la enfermedad la existencia de alteraciones pancreáticas exocrinas y endocrinas tiende a normalizarse tras el tratamiento con corticosteroides (61); sin embargo en las fases avanzadas, con intensa fibrosis, los cambios tienden a ser permanentes (con atrofia pancreática, irregularidades pancreáticas) (37,62). Las manifestaciones extrapancreáticas igualmente ceden cuando la fibrosis no es intensa. Cuando existe colestasis, con el tratamiento con corticoides, hace innecesaria la colocación de stents o su recolocación.

Tras el tratamiento en numerosos pacientes recurren las manifestaciones clínicas y morfológicas, haciendo necesario el tratamiento preventivo con corticoides a dosis reducidas, habitualmente con 2,5 a 10 mg/día, durante largos periodos. Dada la edad avanzada de la mayoría de los pacientes, se ha descrito la aparición de complicaciones secundarias a su uso. A este propósito algunos autores han empezado a utilizar alternativamente inmunomoduladores como la azatioprina como tratamiento postagudo de mantenimiento de la remisión de las complicaciones, con resultados preliminares esperanzadores (63).
Pancreatitis autoinmune. Criterios diagnósticos
No existe un consenso unánime sobre los criterios a utilizar en el diagnóstico de la pancreatitis autoinmune, habiendo sido publicados diferentes en el curso de pocos años (5,19,23,25,39,40,64) y que, dada la rapidez con la que se van modificando los conocimientos sobre la enfermedad, unos van quedando superados por otros en pocos años.
En la actualidad se utilizan principalmente los criterios de la escuela japonesa (40), los de la escuela de la Clínica Mayo (25), además de los propuestos por Frulloni y cols. en Italia (5) y Kim y cols. en Corea (39).
En 2006, en Japón se llega a un acuerdo de mínimos sobre los criterios clínicos diagnósticos de la pancreatitis autoinmune (40), sustituyendo a los propuestos unos años antes por la Japan Pancreatic Society (64). En estos criterios diagnósticos se utilizan los datos correspondientes a las manifestaciones morfológicas de imagen, biológicas e histopatológicas (Tabla XII). Recomiendan la CPRE para un adecuado diagnóstico de las alteraciones del conducto pancreático (Tabla IX) y consideran que con la colangiopancreatografía por RNM se dan dificultades interpretativas. Entre los criterios diagnósticos no se incluye la apreciación de una masa tumefactiva en el páncreas ni la asociación con la colitis ulcerosa, como se ha indicado entre los autores occidentales (5). Se considera que el conducto pancreático está característicamente estrechado en más de 1/3 de su longitud, a diferencia de lo que habitualmente se observa en otras pancreatopatías. Cuando las pruebas de imagen son típicas pero las serológicas son negativas, puede darse la pancreatitis autoinmune, siendo en este caso necesaria la comprobación histopatológica. Se indica que, para el estudio histopatológico, el tejido obtenido mediante biopsia con aguja fina guiada ecoendoscópicamente puede ofrecer grandes dificultades diagnósticas si la muestra es demasiado pequeña.
También en el año 2006 Chari y cols. exponen los criterios de la Clínica Mayo (Tabla XIII) (25), con diferencias significativas con respecto los propuestos por la escuela japonesa. Se consideran tres categorías diagnósticas según diferentes situaciones clínicas: a) aquellas en las que el diagnóstico se basa en datos histopatológicos de pancreatitis linfoplasmática esclerosante (11,65); b) las que se basa en datos de imagen típicas con aumento sérico de IgG4; y c) las que se basan en la presencia de una pancreatopatía idiopática con aumento sérico de IgG4 y/o demostración de la afectación histopatológica típica de otros órganos, con respuesta clara tras el tratamiento con corticosteroides. Estos autores también cuestionan el valor aislado de las biopsias por punción (core biopsias) por la dificultad en la obtención de suficiente tejido como para poder caracterizar adecuadamente las alteraciones histopatológicas descritas con las técnicas histológicas clásicas, y en estos casos se considera que es necesario el estudio inmunohistológico con demostración de la infiltración significativa de plasmacélulas IgG4 + (63). A diferencia de los criterios japoneses, el grupo de la Clínica Mayo introduce el parámetro de la respuesta al tratamiento con corticosteroides, con la advertencia de que no debe utilizarse hasta conseguida la exclusión de otras pancreatopatías y la existencia de alteraciones objetivables donde se pueda confirmar claramente la respuesta terapéutica. Estos autores observan que esta prueba adecuadamente utilizada es positiva en el 100% de sus pacientes, haciendo hincapié en el hecho de que el tratamiento con los corticoides va a modificar únicamente el componente infiltrativo linfoplasmocitario, pero no la intensa fibrosis causante de alteraciones permanentes tanto morfológicas como funcionales (atrofia pancreática). Según estos autores, aunque el diagnóstico histopatológico constituye la prueba de oro de esta enfermedad, este no se consigue adecuadamente en el 41% de sus casos, utilizando entonces uno de los otros dos criterios (Tabla XIII) con los que califica adecuadamente el 100% de sus casos (25).
Evolución a largo plazo
No son bien conocidos la evolución y el pronóstico de esta enfermedad. Las series publicadas comprenden periodos de tiempo limitados con un número reducido de pacientes (62,66-69).
Algunos pacientes mejoran espontáneamente (68,70-72), aunque esta mejoría se consigue habitualmente tras el tratamiento con corticoides (25,39,40).
En las fases iniciales las alteraciones pancreáticas funcionales exocrinas y endocrinas revierten tras este tratamiento (61), como corresponde a los hallazgos patológicos, donde predomina la infiltración linfoplasmocitaria (73), pero en más del 50% de los casos, generalmente en fase avanzada, la diabetes y la esteatorrea secundarias a la atrofia pancreática con masiva fibrosis son irreversibles (37,43,73).
Se ha observado que en los pacientes tratados con corticoides la recurrencia de los síntomas se ha producido del 10 al 40% de los casos, necesitando volver a tratar (67) y en algunos casos ha obligado a mantener el tratamiento con dosis más reducidas (37,63,67-69).
Recientemente Hirano y cols. (67), en un estudio retrospectivo sobre pacientes tratados con corticosteroides (16 casos) y sin este tratamiento (19 casos), constatan que tras un seguimiento medio de 25 meses los pacientes sin tratamiento habían desarrollado diferentes incidentes desfavorables en el 70% de ellos, en forma de ictericia obstructiva, molestias abdominales, colangitis, fibrosis retroperitoneal, nefritis e hidronefrosis. En los enfermos tratados con corticosteroides los diferentes incidentes desfavorables se redujeron al 32%. En algunos casos se observa la aparición de complicaciones secundarias al tratamiento prolongado con corticosteroides, como fractura vertebral, necrosis avascular femoral, descompensación de la diabetes o cataratas, etc.
Al no estar normalizado el concepto de recurrencia (de los síntomas como ictericia, dolor abdominal, de las alteraciones de imagen pancreáticas o extrapancreáticas, alteraciones exocrinas/endocrinas del páncreas, etc.) así como de las dosis de costicosteroides, tanto al inicio como de mantenimiento (63), las conclusiones de estos trabajos, generalmente retrospectivos, son sólo orientativas.
En contra de lo considerado inicialmente, con la evolución pueden aparecer cálculos pancreáticos, especialmente cuando los pacientes presentan recurrencias, lo que hace suponer que finalmente estos pacientes evolucionan hacia lo que constituye una pancreatitis crónica "clásica" (66).
Algunos autores han observado en el seguimiento la aparición de neoplasias, tanto de origen pancreático como extrapancreático, en un número reducido de casos -3 de 42 en la serie de Hirano y cols. (67) y 2 de 10 en la serie de Nishino y cols. (68)-, lo que obliga a analizar con cautela la evolución de estos pacientes.
Bibliografía
1. Sarles H, Sarles JD, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas -An autonomous pancreatic disease? Am J Dig Dis 1961; 6: 688-98. [ Links ]
2. Oliver-Pascual E, Sanz-Ibáñez J, Fernández E, Puras E. Pancreatitis crónica por esclerosis total del páncreas, clínicamente latente y cirrosis biliar de origen extrahepático. Relación de ambos procesos. Rev Esp Enferm Ap Digest Nutric 1961; 20: 287-300. [ Links ]
3. Sarles H, Adler G, Dani R, Frey C, Gullo L, Harada H. The classification of pancreatitis and definition of pancreatic diseases. Digestion 1989; 43: 34-6. [ Links ]
4. Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by autoimmune abnormality. Proposal of concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40: 1561-8. [ Links ]
5. Pearson RK, Longnecker DS, Chari ST, Smyrk TC, Okazaki K, Frulloni L, et al. Controversies in clinical pancreatology: Autoimmune pancreatitis: Does it exist?. Pancreas 2003; 27: 1-13. [ Links ]
6. Kawaguchi K, Koike M, Tsuruta K, Okamoto A, Tabata I, Fujita N. Lymphoplasmacytic sclerosing pancreatitis with cholangitis: A variant of primary sclerosing cholangitis extensively involving pancreas. Hum Pathol 1991; 22: 387-95. [ Links ]
7. Motoo Y, Minamoto T, Watanabe H, Sakai J, Okai T, Sawabu N. Sclerosing pancreatitis showing rapidly progressive changes with recurrent mass formation. Int J Pancreatol 1997; 21: 85-90. [ Links ]
8. Ectors N, Maillet B, Aerts R, Geboes K, Donner A, Borchard F, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41: 63-8. [ Links ]
9. Erkelens GW, Vleggaar FP, Lesterhuis W, van Buuren HR, van der Werf SD. Sclerosing pancreato-cholangitis responsive to steroid therapy. Lancet 1999; 354: 43-4. [ Links ]
10. Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: Clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27: 1119-7. [ Links ]
11. Yadav D, Notahara K, Smyrk T, Clain J, Pearson R, Farnell M, et al. Idiopathic tumefactive chronic pancreatitis: Clinical profile, histology, and natural history after resection. Clin Gastroenterol Hepatol 2003; 1: 129-35. [ Links ]
12. Kodama T, Abe M, Sato H, Imamura Y, Koshitani T, Kato K, et al. A case of pseudotumorous pancreatitis that presented unique pancreatoscopic findings with the peroral electronic pancreatoscope. J Gastroenterol Hepatol 2003; 18: 108-11. [ Links ]
13. Kamisawa T, Okamoto A. Autoimmune pancreatitis: Proposal of IgG4-related sclerosing disease. J Gastroenterol 2006; 41: 613-5. [ Links ]
14. Kamisawa T. IgG4-positive plasma cells specifically infiltrate various organs in autoinmmune pancreatitis. Pancreas 2004; 29: 167-8. [ Links ]
15. Frulloni L, Bovo P, Brunelli S, Vaona B, Di Francesco V, Nishimori I, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20: 382-8. [ Links ]
16. Nishimori I, Yamamoto Y, Okazaki K, Morita M, Onodera M, Kino J, et al. Identification of autoantibodies to a pancreatic antigen in patients whit idiopathic pancreatitis and Sjögren's syndrome. Pancreas 1994; 9: 374-81. [ Links ]
17. Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54: 274-81. [ Links ]
18. Kino-Ohsaki J, Nishimori I, Morita M, Okazaki K, Yamamoto Y, et al. Serum antibodies to carbonic anhydrase I and II in patients with idiopathic chronic pancreatitis and Sjögren's syndrome. Gastroenterology 1996; 110: 1579-86. [ Links ]
19. Aparisi L, Farré A, Gómez-Cambronero L, Martínez J, de las Heras G, Corts J, et al. Antibodies to carbonic anhydrase and IgG4 levels in idiopathic chronic pancreatitis: Relevance for diagnosis of autoimmune pancreatitis. Gut 2005; 54: 703-9. [ Links ]
20. Okazaki K, Uchida K, Ohana M, Nakase H, Uose S, Inai M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th -type cellular immune response. Gastroenterology 2000; 118: 573-81. [ Links ]
21. Uchida K, Okazaki K, Nishi T, Uose S, Nakase H, Ohana M, et al. Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Lab Invest 2002; 82 : 411-24. [ Links ]
22. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344: 732-8. [ Links ]
23. Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355: 2670-6. [ Links ]
24. Kawa S, Ota M, Yoshizawa K, Horiuchi A, Hamano H, Ochi Y, et al. HLA DRB1*0405-DQB1 Haplotype is associated with autoimmune pancreatitis in Japanese population. Gastroenterology 2002; 122: 1264-9. [ Links ]
25. Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4: 1010-6. [ Links ]
26. Uchida K, Okazaki K, Konishi Y, Ohana M, Takakuwa H, Hajiro K, et al. Clinical analysis of autoimmune-related pancreatitis. Am J Gastroenterol 2000; 95: 2788-94. [ Links ]
27. Okazaki K. Autoimmune pancreatitis is increasing in Japan. Gastroenterology 2003; 125: 1557-8. [ Links ]
28. Kim KP, Kim MH, Song MH, Lee SS, Seo DW, Lee SK. Autoimmune chronic pancreatitis. Am J Gastroenterol 2004; 99: 1605-16. [ Links ]
29. Ghazale A, Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, et al. Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102: 1646-53. [ Links ]
30. Weber SM, Cubukcu-Dimopulo O, Palesty JA, Suriawinata A, Klimstra D, Brennan MF, et al. Lymphoplasmacytic sclerosing pancreatitis: Inflammatory mimic of pancreatic carcinoma. J Gastrointest Surg 2003; 7: 129-37. [ Links ]
31. Zamboni G, Lüttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: A study on 53 resection specimens and 9 biopsy specimens. Virchow Arch 2004; 445: 552-63. [ Links ]
32. Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A, Kamata N. Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma. Am J Gastroenterol 2003; 98: 2694-9. [ Links ]
33. Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol 2006; 41: 1197-05. [ Links ]
34. Ito T, Kawabe K, Arita Y, Hisano T, Igarashi H, Funakoshi A, et al. Evaluation of pancreatic endocrine and exocrine function in patients with autoimmune pancreatitis. Pancreas 2007; 34: 254-9. [ Links ]
35. De las Heras-Castaño G, Castro-Senosiain B, García-Suárez C, López-Hoyos M, San-Segundo D, Juanco C, et al. Pancreatitis autoinmune. ¿Una entidad poco frecuente o infradiagnosticada?. Características anatomopatológicas, clínicas e inmunológicas. Gastroenterol Hepatol 2006; 29: 299-305. [ Links ]
36. Nishimori I, Tamakoshi A, Kawa S, Tanaka S, Takeuchi K, Kamisawa T, et al. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: Findings from a nationwide survey in Japan. Pancreas 2006; 32: 244-8. [ Links ]
37. Beristain JL, Sabater L, Calatayud A, Calvete J, Rausell M, Lledó S, et al. Pancreatitis autoinmune: pseudotumor inflamatorio, afectación multifocal y evolución a largo plazo. Rev Esp Enfem Dig (en prensa). [ Links ]
38. Hirano K, Kawabe T, Yamamoto N, Nakai Y, Sasahira N, Tsujino T, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367: 181-4. [ Links ]
39. Kim KP, Kim MH, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12: 2487-96. [ Links ]
40. Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishimori I, et al. Clinical diagnostic criteria of autoimmune pancreatitis: Revised proposal. J Gastroenterol 2006; 41: 626-31. [ Links ]
41. Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 7 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55: 494-9. [ Links ]
42. Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Okai T, et al. Clinical and imaging features of autoimmune pancreatitis with focal pancreatic swelling or mass formation: Comparison with so-called tumor-forming pancreatitis and pancreatic carcinoma. Am J Gastroenterol 2003; 98: 2679-87. [ Links ]
43. Nishimori I, Bratanova T, Toshkov I, Caffrey T, Mogaki M, et al. Induction of experimental autoimmune sialoadenitis by immunization of PL/J mice with carbonic anhydrase II. J Immunol 1995;154: 4865-73. [ Links ]
44. Sahani DV, Kalva SP, Farrell J, Maher MM, Saini S, Mueller PR, et al. Autoimmune pancreatitis: Imaging features. Radiology 2004; 233: 345-52. [ Links ]
45. Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Morphological changes after steroid therapy in autoimmune pancreatitis. Scand J Gastroenterol 2004; 39: 1154-8. [ Links ]
46. Kamisawa T, Chen P, NakajimaY , Egawa N, Tsuruta K, Okamoto A, et al. Pancreatic cancer with a high serum IgG4 concentration. World J Gastroenterol 2006; 12: 6225-8. [ Links ]
47. Kamisawa T, Chen PY, Tu Y, Nakajima H, Egawa N, Tsuruta K, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12 (18): 2919-22. [ Links ]
48. Kamisawa T, Funata N, Hayashi Y, Tsuruta T, Okamoto A, Amemiya K, et al. Close relationship between autoimmune pancreatitis and multifocal fibroesclerosis. Gut 2003; 52: 683-7. [ Links ]
49. Nishino T, Toki F, Oyama H, Oi I, Kobayashi M, Takasaki K, et al. Biliary tract involvement in autoimmune pancreatitis. Pancreas 2005; 30: 76-82. [ Links ]
50. Ohara H, Nakazawa T, Ando T, Joh T. Systemic extrapancreatic lesions associated with autoimmune pancreatitis. J Gastroenterol 2007; 42 (Supl. 18): 15-21. [ Links ]
51. Nishino T, Oyama H, Hashimoto E, Toki F, Oi I, Kobayashi M, et al. Clinicopathological differentiation between sclerosing cholangitis with autoimmune pancreatitis and primary sclerosing cholangitis. J Gastroenterol 2007; 42: 550-9. [ Links ]
52. Kamisawa T, Chen PY, Tu Y, Nakajima H, Egawa N. Autoimmune pancreatitis metachronously associated with retroperitoneal fibrosis with IgG4-positive plasma cell infiltration. World J Gastroenterol 2006; 12: 2955-7. [ Links ]
53. Deshpande V, Chicano S, Finkelberg D, Selig MK, Mino-Kenudson M, Brugge WR, et al. Autoimmune pancreatitis: A systemic immune complex mediated disease. Am J Surg Pathol 2006; 30: 1537-45. [ Links ]
54. Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology 2007; 242: 791-801. [ Links ]
55. Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant 2004; 19: 474-6. [ Links ]
56. Shinji A, Sano K, Hamano H, Unno H, Fukushima M, Nakamura N, et al. Autoimmune pancreatitis is closely associated with gastric ulcer presenting with abundant IgG4-bearing plasma cell infiltration. Gastrointest Endosc 2004; 59: 506-11. [ Links ]
57. Ohara H, Nakazawa T, Sano H, Ando T, Okamoto T, Takada H, et al. Systemic extrapancreatic lesions associated with autoimmune pancreatitis. Pancreas 2005; 31: 232-7. [ Links ]
58. Zhang L, Notohara K, Levy MJ, Chari ST, Smyrk TC. IgG4-positive plasma cell infiltration in the diagnosis of autoimmune pancreatitis. Mod Pathol 2007; 20: 23-8. [ Links ]
59. Bang S, Kim M, Kim D, Lee T, Kwon S, Oh H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36: 84-9. [ Links ]
60. Chari ST. Current concepts in the treatment of autoimmune pancreatitis. J Pancreas 2007; 8: 1-3. [ Links ]
61. Kamisawa T, Egawa N, Inokuma S, Tsuruta K, Okamoto A, Kamata N, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27: 235-8. [ Links ]
62. Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: Results from a long-term follow-up study. Pancreatology 2005; 5: 234-8. [ Links ]
63. Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56: 1650-2. [ Links ]
64. Members of the Criteria Committee for Autoimmune Pancreatitis of the Japan Pancreas Society. Diagnostic criteria for autoimmune pancreatitis by the Japan Pancreas Society. J Jpn Pan Soc 2002; 17: 585-7. [ Links ]
65. Klöppel G, Lüttges J, Löhr M, Zamboni G, Longnecker D. Autoimmune pancreatitis: Pathological, clinical, and immunological features. Pancreas 2003; 27: 14-9. [ Links ]
66. Kamisawa T, Chung JB, Irie H, Nishino T, Ueki T, Takase M, et al. Japan-Korea symposium on autoimmune pancreatitis (KOKURA 2007). Pancreas 2007; 35: 281-4. [ Links ]
67. Hirano K, Tada M, Isayama H, Yagioka H, Sasaki T, Kogure H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56: 1719-4. [ Links ]
68. Nishino T, Toki F, Oyama H, Shimizu K, Shiratori K. Long-term outcome of autoimmune pancreatitis after oral prednisolone therapy. Intern Med 2006; 45: 497-501. [ Links ]
69. Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: Strategy for steroid treatment. Pancreas 2005; 30: 31-9. [ Links ]
70. Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42 (Supl. 18): 59-62. [ Links ]
71. Kubota K, Iida H, Fujisawa T, Yoneda M, Inamori M, Abe Y, et al. Clinical factors predictive of spontaneous remission or relapse in cases of autoimmune pancreatitis. Gastrointest Endosc 2007; 66 (6): 1142-51. [ Links ]
72. Ozden I, Dizdaroðlu F, Poyanli A, Emre A. Spontaneous regression of a pancreatic head mass and biliary obstruction due to autoimmune pancreatitis. Pancreatology 2005; 5 (2-3): 300-3. [ Links ]
73. Suda K, Nishimori I, Takase M, Oi I, Ogawa M. Autoimmune pancreatitis can be classified into early and advanced stages. Pancreas 2006; 33: 345-50. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Luis Aparisi Quereda.
Servicio de Aparato Digestivo.
Hospital Clínico de Valencia.
Avda. Blasco Ibáñez, 17.
46010 Valencia.
e-mail: aparisi_lui@gva.es
Recibido: 19-02-08.
Aceptado: 25-02-08.

