










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.100 no.10 Madrid oct. 2008
Pancreatitis autoinmune: pseudotumor inflamatorio, afectación multifocal, hipertensión portal y evolución a largo plazo
Autoimmune pancreatitis: inflammatory pseudotumor, multifocal fibrosclerosis, portal hypertension, and long-term outcome
J. L. Beristain, L. Sabater, A. Calatayud1, J. Calvete, M. Rausell2, S. Lledó, J. Tosca3, J. Sastre4 y L. Aparisi
Servicios de Cirugía General y Aparato Digestivo, 1Anatomía Patológica, 2Radiodiagnóstico, 3Hepatología y 4Departamento Fisiología. Hospital Clínico Universitario de Valencia y Universidad de Valencia
Dirección para correspondencia
RESUMEN
La pancreatitis autoinmune es una enfermedad recientemente caracterizada y que en la actualidad constituye un reto diagnóstico especialmente su diferenciación con el cáncer de páncreas. Su evolución a largo plazo es poco conocida, presentándose un caso estudiado a lo largo de 14 años y mostrando su evolución clínica, bioquímica y morfológica.
Paciente mujer de 54 años que debuta con un cuadro de ictericia obstructiva y molestias abdominales inespecíficas y constatación en la TAC de un aumento de la cabeza del páncreas, todo ello sugestivo de neoplasia de páncreas. Fue intervenida evidenciándose un aumento difuso de todo el páncreas descartándose malignidad intraoperatoriamente, realizando únicamente colecistectomía y coledocoduodenostomía, quedando diagnosticada entonces como pancreatitis crónica. Durante los años posteriores fueron apareciendo diferentes procesos autoinmunes como asma, sialoadenitis y colangitis esclerosante secundaria, así como episodios recurrentes de ictericia e insuficiencia pancreática endocrina y exocrina. La aparición de estas complicaciones y la detección de niveles séricos elevados de IgG4 y de anticuerpos antianhidrasa carbónica II condujo a la reevaluación de la histología inicial concluyendo finalmente con el diagnóstico de pancreatitis autoinmune al evidenciarse una infiltración linfocitaria y plasmacitaria IgG4+, así como fibrosis y flebitis obliterativa. En los últimos años se ha añadido a las anteriores complicaciones una fibrosis retroperitoneal con hipertensión portal, varices esofágicas y esplenomegalia.
Palabras clave: Pancreatitis autoinmune. Pancreatitis crónica. IgG4. Plasmacélulas IgG4+. Sialoadenitis. Colangitis esclerosante. Fibrosis retroperitoneal. Hipertensión portal.
ABSTRACT
Autoimmune pancreatitis is a recently characterized disease that still constitutes a diagnostic challenge, especially regarding differential diagnosis from neoplasia. Long-term outcome is poorly known. We herein report a case of a patient with autoimmune pancreatitis and 14 years of follow-up, and show its clinical, biochemical, and morphological characteristics.
A 54-year-old female presented with obstructive jaundice and abdominal tenderness, as well as a mass at the pancreatic head on a CT scan, suggestive of pancreatic neoplasia. Surgery showed an increase of the whole pancreas, malignancy was intraoperatively ruled out, and a cholecystectomy and choledochoduodenostomy were carried out. The diagnosis was chronic pancreatitis. Over the following years different autoimmune complications developed, including asthma, salivary gland swelling, and sclerosing cholangitis, as well as recurrent episodes of jaundice, and exocrine and endocrine pancreatic failure. The development of these complications combined with the demonstration of high serum levels of IgG4 and carbonic anhydrase II led to a re-evaluation of the initial histology of the pancreas, leading to a final diagnosis of autoimmune pancreatitis: IgG4+ lymphoplasmacytic infiltrates, fibrosis, and obliterative phlebitis. New complications developed during the last few years: retroperitoneal fibrosis with portal hypertension, esophageal varices, and splenomegaly.
Key words: Autoimmune pancreatitis. Chronic pancreatitis. IgG4. IgG4-positive plasma cells. Salivary gland swelling. Sclerosing cholangitis. Retroperitoneal fibrosis. Portal hypertension.
Introducción
La pancreatitis autoinmune (PA) es un tipo particular de pancreatitis crónica descrito inicialmente por Sarles en 1961 (1) y posteriormente clínicamente caracterizada por Yoshida en 1995 (2). Los conocimientos sobre esta enfermedad son aún limitados y sólo recientemente ha pasado a considerarse como una enfermedad sistémica fibro-inflamatoria que afecta principalmente al páncreas, además de a otros órganos como los conductos biliares, glándulas salivares, retroperitoneo y ganglios linfáticos (3,4), que presentan específicamente una infiltración linfoplasmocitaria con abundantes células IgG4 positivas (5). Estos pacientes presentan manifestaciones clínicas, de laboratorio, histológicas y de imagen particulares: sintomatología leve e inespecífica, asociación con otras enfermedades autoinmunes, aumento de los niveles séricos de gammaglobulina, especialmente IgG4, presencia de diferentes autoanticuerpos, aumento variable del tamaño del páncreas, estrechamiento irregular del conducto pancreático principal y ocasionalmente del colédoco y finalmente una buena respuesta al tratamiento con corticosteroides (6-8).
La importancia clínica de esta enfermedad radica en que con frecuencia plantea un problema de diagnóstico diferencial con el cáncer de páncreas, en que es más frecuente de lo que se pensaba inicialmente y en la buena respuesta al tratamiento corticoideo.
La prevalencia entre las series médicas oscila entre 2 y 8% de todos los pacientes con pancreatitis crónica (4,9), alcanzando en las series quirúrgicas hasta un 26% (10,11). Es necesario recalcar que la disparidad encontrada en la incidencia entre unas y otras reside principalmente en que las series quirúrgicas encuentran un mayor número de ellas debido al material obtenido para estudio histopatológico durante los procedimientos quirúrgicos.
Aunque en la literatura reciente existen numerosos trabajos sobre los diferentes aspectos clínicos y diagnósticos de la pancreatitis autoinmune, muy pocos presentan el seguimiento a largo plazo de casos de pancreatitis crónica autoinmune mostrando los cambios que acontecen a lo largo de la historia natural de la enfermedad.
En este trabajo se describe una paciente con pancreatitis autoinmune y su evolución clínica, bioquímica y morfológica a lo largo de 14 años. La relevancia del caso estriba en la descripción de las diferentes complicaciones que ha ido presentando a lo largo de los años con su correspondiente estudio histopatológico, así como las diferentes estrategias diagnósticas y terapéuticas que se han realizado durante su evolución. Entre las complicaciones que ha presentado se describe por primera vez la aparición de hipertensión portal con varices esofágicas en un paciente con pancreatitis crónica autoinmune.
Caso clínico
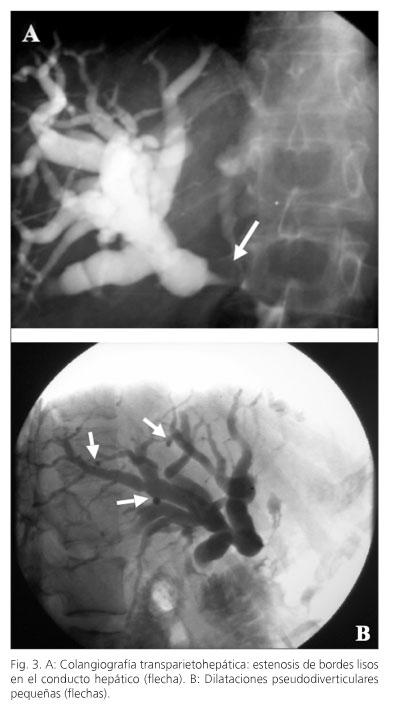
Paciente mujer de 54 años, sin hábitos tóxicos y con el único antecedente de histerectomía por prolapso uterino 3 años antes. Consulta en septiembre de 1993 por ictericia y molestias abdominales inespecíficas, destacando en la analítica una bilirrubina total de 18 mg/dl, con directa de 17 mg/dl y serología para hepatitis B y C negativa. En el estudio por imagen con TAC se observa un aumento focal de la cabeza del páncreas, de 3 cm, homogéneo, de aspecto sólido y dilatación de la vía biliar intra y extrahepática, todo ello sugestivo de neoplasia pancreática (Fig. 1). Se intervino quirúrgicamente, evidenciando una glándula pancreática aumentada de tamaño en toda su extensión, indurada, tomando múltiples biopsias del tejido pancreático y adenopatías peripancreáticas, todas ellas indicativas de pancreatitis crónica. Al no identificar celularidad maligna se realizó colecistectomía y coledocoduodenostomía. En el estudio histológico definitivo se evidenciaron áreas de parénquima pancreático delimitadas por extensas bandas fibrosas con pequeños nidos celulares y ductos atrapados en su seno y reacción inflamatoria mononuclear linfoplasmocitaria folicular y difusa (Fig. 2), por lo que finalmente fue diagnosticada de pancreatitis crónica idiopática.


Durante los siguientes dos años la paciente estuvo asintomática, manteniéndose las alteraciones observadas en las TAC iniciales y desde un punto de vista funcional con curva de glucemia y grasas en heces normales. A lo largo de 1995 persiste en la TAC un páncreas aumentado de tamaño y se realiza una nueva exploración funcional del páncreas, ahora mediante el test de secretina-pancreocimina, que demostró una insuficiencia pancreática moderada, sin esteatorrea. Esta situación empeoró un año después al aparecer esteatorrea (15 g/24 h) y diabetes mellitus, iniciándose tratamiento sustitutivo con enzimas pancreáticas e insulina. En abril de 1997 reingresa por nuevo cuadro de ictericia, dolor abdominal y pérdida de peso, realizándose colangiografía transparietohepática demostrando estrechamiento a nivel de la anastomosis, sugestiva de estenosis fibrosa benigna, así como pequeñas dilataciones pseudodiverticulares en los conductos biliares intrahepáticos (Fig. 3). Se reintervino realizando resección de la vía biliar y una doble hepático-yeyunostomía en Y de Roux. El estudio histopatológico evidenció proceso inflamatorio crónico con esclerosis de las vías biliares extrahepáticas, abundante celularidad inflamatoria, congestión capilar e hiperplasia de pequeños ductos biliares, con discretas atipias citológicas y arquitecturales, sugestivo de colangitis esclerosante (Fig. 4A). Durante el siguiente año se desarrolló un progresivo aumento de las glándulas parótidas sin llegar a un diagnóstico claro del origen de esta sialoadenitis a pesar de la biopsia y con una sialografía en la que no se observaban litiasis. En esos momentos analíticamente se constató la presencia sérica de anticuerpos antinucleares positivos, y anti-mitocondriales, anti-músculo liso y anti-RO (Ssa) negativos, además de una marcada eosinofilia (3,4 x 109/l, 32%) siendo el estudio de parásitos en heces negativo por lo que se remitió para estudio de un posible proceso alérgico que resultó también negativo. En 1998 ingresa por disnea, ictericia y síndrome constitucional, siendo catalogado el cuadro respiratorio de asma bronquial y tratado con broncodilatadores y metilprednisolona. En cuanto al cuadro de ictericia fue evaluado mediante pruebas de imagen y biopsia hepática confirmándose el diagnóstico de colangitis esclerosante (Fig. 4B). A los pocos días de iniciado el tratamiento corticoideo desaparecen la disnea, la ictericia y el cuadro constitucional de forma espectacular. Tras el alta hospitalaria fueron retirados los corticoides, manteniéndose como tratamiento ácido ursodesoxicólico, enzimas pancreáticas e insulina. En aquel momento la IgG total fue de 2.026 mg/dl (normal < 1.600 mg/dl), lo que unido a la hiperreactividad bronquial, sialoadenitis y colangitis esclerosante hizo suponer la existencia de un proceso autoinmune. Asintomática durante los siguientes 2 años, aparece entonces un síndrome de Sjögren. En 2002 y estando asintomática se realizó una RNM de control que mostró un páncreas atrófico con un Wirsung de calibre filiforme y permeabilidad de la vena porta y sus ramas, además de una importante esplenomegalia y ocupación retroperitoneal por una masa polilobulada que engloba al tronco celiaco que finalmente se diagnostica como fibrosis retroperitoneal. Se realizó además un test de aclaramiento hepático de verde de indocianina con unos valores en el límite de la normalidad (PDR = 19,6 %/min; N > 20). En el control analítico de diciembre de 2003 se constató el progresivo aumento sérico de las IgG (3.812 mg/dl), la notable elevación de las IgG4 (23.200 mg/l, normal < 1.300 mg/l) y de los anticuerpos anti-anhidrasa carbónica II. Todos estos resultados motivaron la revisión histopatológica e inmunohistoquímica de las piezas que se conservaban en parafina de las intervenciones y biopsias previas, demostrándose la presencia de un intenso infiltrado linfocitario y de plasmocitos IgG4+ característico de pancreatitis autoinmune en el páncreas y en diferentes órganos (Fig. 5). En junio de 2004 sufre una nueva crisis asmática que fue tratada con prednisolona y con posterioridad ese mismo año un nuevo episodio de empeoramiento clínico con astenia, pérdida de peso y distensión abdominal por lo que se efectúa una TAC en la que se observa un páncreas atrófico con conducto pancreático filiforme, esplenomegalia, signos de hipertensión portal con varices esofágicas, aumento de la pared gástrica y fibrosis retroperitoneal (Fig. 6). Se rea-lizó una fibrogastroscopia en la que se observaron varices esofágicas grado IV y gastropatía congestiva. La biopsia gástrica constató una marcada infiltración linfoplasmocitaria y en la analítica se pudo demostrar una IgG total de 6.730 mg/dl, IgG4 30.200 mg/l y plaquetopenia. Se inició tratamiento con prednisona 40 mg/día durante 4 semanas con reducción progresiva y mejorando el cuadro clínico y reducción de las inmunoglobulinas (IgG total 1.325 mg/dl e IgG4 9.400 mg/l) (Fig. 7). Desde entonces fue presentando cuadros ocasionales de astenia y pérdida de peso que mejoraban con la reintroducción de corticoides y que finalmente ha obligado en la actualidad a mantener una dosis diaria de 10 mg/día. La exploración hepática más reciente mediante elastografía (Fibroscan) muestra un resultado de 16,6 kPa (N < 7,9), indicativo de fibrosis significativa, y un nuevo test de verde de indocianina alcanza un valor semejante al obtenido 3 años antes (PDR = 19,6%/min). En la actualidad, a sus 68 años la paciente se encuentra con un razonable buen estado general teniendo en cuenta su insuficiencia exocrina y endocrina pancreática y sin precisar reingresos desde hace 3 años.


Discusión
La pancreatitis autoinmune comprende un complejo de manifestaciones clínicas, biológicas, morfológicas y de respuesta terapéutica. Entre las características clínicas destaca la colestasis, la ausencia o leve dolor pancreático y la asociación con otras afecciones autoinmunes; entre las biológicas, el aumento sérico de gammaglobulinas y especialmente la IgG4, así como la presencia de diferentes autoanticuerpos (ANA, anti-anhidrasa carbónica-II); en cuanto a las alteraciones morfológicas de imagen señalar el aumento focal o difuso del páncreas (características pseudotumorales), disminución irregular segmentaria o difusa del conducto pancreático, y la infrecuente aparición de calcificaciones o pseudoquistes pancreáticos; finalmente se considera también indicativo de esta enfermedad su buena respuesta al tratamiento con corticoides. Sin embargo, el diagnóstico definitivo viene dado por la demostración a nivel de tejido pancreático de una intensa infiltración periductal linfocitaria y de células plasmáticas IgG4+, una marcada fibrosis interlobular y periductal y flebitis obliterativa (3,6,11,12).
Uno de los aspectos más importantes al considerar la pancreatitis autoinmune es su diagnóstico diferencial con el cáncer de páncreas ya que con frecuencia se presenta como un cuadro de colostasis poco dolorosa asociado o no a una masa o pseudotumor a nivel de la cabeza pancreática. De hecho, la mayoría de los trabajos publicados en los últimos años son trabajos retrospectivos sobre sujetos intervenidos por sospecha de neoplasia pancreática (13), aunque se han propuesto una serie de criterios diferenciales tanto por la escuela japonesa (12) como por los investigadores de la Clínica Mayo (3).
El caso presentado se interpretó inicialmente como un cáncer de páncreas. Fue intervenido quirúrgicamente y se descartó la neoplasia con el estudio histopatológico de múltiples muestras tomadas en la operación, siendo caracterizada de pancreatitis crónica con infiltración linfoplasmocitaria que en su momento, 1993, no se supo interpretar al no estar todavía definidas las características histopatológicas de la enfermedad. Durante los años siguientes la paciente fue presentando diferentes episodios clínicos y manifestaciones que apuntaban hacia un proceso autoinmune (colangitis esclerosante, sialoadenitis, asma bronquial, síndrome de Sjögren), teniendo en cuenta además que mejoraban con la administración de corticoides. La última de estas manifestaciones, la fibrosis retroperitoneal, se acompañaba a su vez de hipertensión portal con varices esofágicas y esplenomegalia. La hipertensión portal y especialmente la aparición de varices esofágicas no han sido descritas hasta ahora en la literatura asociadas a la pancreatitis autoinmune. Como causa de esta complicación se puede considerar por un lado la fibrosis retroperitoneal y por otro la fibrosis portal intrahepática existente. La fibrosis retroperitoneal ha sido observada en el 12,5% de los casos de la serie de Hamano provocando en alguno de ellos alteración hidronefrótica por afectación de las vías urinarias (14,15).
En 2001, Hamano y cols. (8) demuestran que la fracción 4 de las inmunoglobulinas IgG tenían un gran valor diagnóstico en la pancreatitis autoinmune, lo que nos ayudó posteriormente a perfilar el diagnóstico en nuestra paciente. Más recientemente, Ghazale y cols. (9) han obtenido, para un punto de corte de 1.400 mg/L de IgG4 sérica, una sensibilidad del 76% y una especificidad del 93% en el diagnóstico de esta enfermedad.
Sin embargo, la confirmación definitiva en nuestra paciente sólo se consiguió al reevaluar las piezas de tejido pancreático obtenido en la primera intervención de 1993 y que se conservaban en parafina, demostrando la presencia de abundantes plasmocitos IgG4+ siguiendo la metodología publicada en 2003 por Kamisawa (14). Nuestro caso tiene también interés al constatar la afectación multifocal de la enfermedad demostrada histológicamente a nivel de páncreas, vías biliares, estómago, intestino delgado e hígado, a lo que hay que añadir la evidencia clínica de la implicación de las glándulas salivales y lacrimales.
Finalmente, uno de los criterios que se han utilizado para el diagnóstico de la pancreatitis autoinmune es la buena respuesta al tratamiento con corticoides (3,12). En nuestra paciente en los diferentes momentos en los que se han utilizado han hecho remitir espectacularmente las manifestaciones clínicas incluyendo la ictericia, el síndrome constitucional y los síntomas respiratorios, así como una evidente disminución de la tasa sérica de IgG4 desde el momento en que se monitorizaron como parte del control analítico sin que en ningún momento hayan llegado a normalizarse (Fig. 7). En la actualidad se mantiene un tratamiento continuado con una dosis de 10 mg de prednisona al día.
Bibliografía
1. Sarles H, Sarles JD, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas-An autonomous pancreatic disease? Am J Dig Dis 1961; 6: 688-98. [ Links ]
2. Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by autoimmune abnormality. Proposal of concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40: 1561-8. [ Links ]
3. Chari S T, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4: 1010-6. [ Links ]
4. Kamisawa T, Okamoto A. Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol 2006; 41: 613-25. [ Links ]
5. Kamisawa T. IgG4-positive plasma cells specifically infiltrate various organs in autoinmmune pancreatitis. Pancreas 2004; 29: 167-8. [ Links ]
6. Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355: 2670-6. [ Links ]
7. Aparisi L, Farre A, Gómez-Cambronero L, Martínez J, De las Heras G, Corts J, et al. Antibodies to carbonic anhydrase and IgG4 levels in idiopathic chronic pancreatitis: relevance for diagnosis of autoimmune pancreatitis. Gut 2005; 54 (5): 703-9. [ Links ]
8. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344: 732-8. [ Links ]
9. Ghazale A, Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, et al. Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102: 1646-53. [ Links ]
10. Yadav D, Notahara K, Smyrk TC. Idiopathic tumefactive chronic pancreatitis: clinical profile, histology, and natural history after resection. Clin Gastroenterol Hepatol 2003; 1: 129-35. [ Links ]
11. Zamboni G, Lüttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchow Arch 2004; 445: 552-63. [ Links ]
12. Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishimori I, et al. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41: 626-31. [ Links ]
13. Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A, Kamata N. Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma. Am J Gastroenterol 2003; 98: 2694-9. [ Links ]
14. Kamisawa T, Funata N, Hayashi Y, Tsuruta T, Okamoto A, Amemiya K, et al. Close relationship between autoimmune pancreatitis and multifocal fibroesclerosis. Gut 2003; 52: 683-7. [ Links ]
15. Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrahepatic lesions complicating autoinmune pancreatitis. J Gastroenterol 2006; 41: 1197-205. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Luis Aparisi Quereda.
Servicio de Hepatología.
Hospital Clínico Universitario de Valencia.
Avda. Blasco Ibáñez, 17.
46010 Valencia.
e-mail: aparisi_lui@gva.es
Recibido: 15-04-08.
Aceptado: 29-04-08.

