










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.101 n.8 Madrid Aug. 2009
Surgical management of hereditary colorectal cancer: surgery based on molecular analysis and family history
Manejo quirúrgico del cáncer colorrectal hereditario: cirugía basadas en el análisis molecular y los antecedentes familiares
J. Perea, I. Justo, E. Álvaro, M. Lomas, J. Díaz Tasende1, J. C. Marín1, A. Franco1, F. Colina2,3, Y. Rodríguez2, J. Martínez4, L. Robles5, M. Urioste6 and M. Hidalgo
Departments of General Surgery B, 1Gastroenterology, 2Pathology, 3Tumor Registry, 4Molecular Biology Unit, 5Genetic Counsel Unit. Department of Oncology. "12 de Octubre" University Hospital. Madrid, Spain. 6Human Genetics Group. Spanish National Cancer Research Center. Madrid, Spain
ABSTRACT
The importance of colorectal cancer (CRC) is increasing. A proportion show a hereditary component, as in Lynch syndrome and Familial Adenomatous Polyposis, and a recently defined entity as well, namely, Familial Colorectal Cancer type X. The high probability to develop CRC in these groups may, at the time of recognition, change surgical management, including its timing or even the surgical technique. In some cases prophylactic surgery can play an important role. The possibility of using tools that allow recognition of the aforementioned syndromes, including microsatellite instability, immunohistochemistry for DNA mismatch repair system proteins, and especially their mutations, is on the basis of therapeutic strategies that differ from those employed in sporadic CRC cases.
Key words: Colorectal cancer. Surgery. Lynch syndrome. Familial adenomatous polyposis. Familial colorectal cancer type X.
Introduction
The incidence of colorectal cancer (CRC) is very high nowadays. Data from the Spanish Health Ministry show that 25,665 new cases were diagnosed in 2003 (1). It is estimated that 20% of patients with CRC have familial aggregation, and around 5% of all cases are hereditary.
CRC with a hereditary component includes Lynch syndrome, or hereditary non-polyposis colorectal cancer (HNPCC), which is also the most common form, and familial adenomatous polyposis (FAP), with two phenotypes, classic and attenuated.
In the last few years a possible new hereditary form of CRC has been defined, called familial colorectal cancer type X. This form meets the Amsterdam criteria for Lynch syndrome, but from a molecular point of view does not follow the same oncogenetic pathway, characterized by the presence of microsatellite instability (MSI) (2-4).
Each of these groups carries some degree of increased risk for CRC development, very high in Lynch syndrome and FAP, and not very well defined in familial CRC type X. Each group needs a different management strategy not only during follow-up, but also regarding the possibility of chemoprevention and of prophylactic surgery to prevent the development of a primary CRC or a metachronic neoplasm. The risk of CRC in FAP is 100% (5), so prophylactic surgery is a necessity in this case. In other forms, as Lynch syndrome, surgery is considered once the CRC is diagnosed because the cumulative risk of metachronic CRC through the age of 60 is nearly 35% (6); notwithstanding this, the risk for a first CRC is 75% (7), depending on multiple factors such as the altered gene and type of mutation. In the other familial forms, in which the risk of developing a CRC remains unknown, the role of prophylactic surgery is yet to be defined, taking into account the major criteria defined by You et al. (8) when addressing prophylactic surgery: presence of genetic tests that allow identification of mutation carriers; a high penetrance associated with the mutation; an effective surgical technique to reduce the risk with tolerable morbidity; an affordable loss of organ function; and the possibility to assess that the patient remains disease-free during follow-up.
The aim of this work is to underline the importance of this group of patients with hereditary syndromes or with CRC aggregation. Their identification may change surgical management: namely, surgical timing as well as the technique performed, and establish the role of prophylactic surgery. We will try to gain some insight into its molecular basis, and also into the possibility of its application to clinical practice.
Material and methods
We present three cases that show the three types of familial CRC. The first and the third ones, examples of Lynch syndrome and FAP, have a defined hereditary component and a clear genetic basis. The remaining one is a potential case of familial CRC type X. In each one, molecular determinations were carried out to determine whether they belong to one of the known syndromes. Surgical management was based on the results of these molecular measurements.
Results
Case 1
A 40-year-old female was diagnosed with CRC of the right colon and evaluated at the High Risk CRC Unit in our institution because of her familial cancer history. Her medical history only showed some plastic surgery procedures for a traumatic lesion, and her familial cancer history fulfilled Amsterdam criteria type I (Fig. 1). Diagnostic colonoscopy showed an ulcerated neoplasm in the right colon; two polyps of the left colon were resected at the same exploration. Staging workup was without findings. Biopsy of the neoplasm confirmed the diagnosis of CRC; the resected polyps were tubular adenomas, without evidence of dysplasia. Immunochemistry showed an absent expression of Msh2 and Msh6 Mismatch Repair System (MMR).

Due to the clinical suspicion of Lynch syndrome, and after obtaining the informed consent, a subtotal colectomy was carried out. The pathology report was of a well-differentiated, mucinous colorectal adenocarcinoma infiltrating pericolic fat, with 28 lymph nodes free of disease, stage B2 (Astler-Coller modified classification). The postoperative course was uneventful and the patient was discharged in good condition. Genetic testing showed a pathogenetic mutation in MSH2 (c.467 G > T, exon 9) thus confirming the diagnosis of Lynch syndrome. This mutation causes a stop signal and a truncated protein. The patient was referred to the Genetic Counsel Unit.
Case 2
A 42-year-old male presented with microcytic and hypochromic anemia. His only previous disease was hepatic cysts. His family cancer history showed that his father was diagnosed with CRC at the age of 64, and a paternal aunt had died from ovarian cancer at the age of 60 (Fig. 2). A colonoscopy and upper digestive endoscopy were carried out, in which a malignant neoplasm at the hepatic flexure of the colon was found and biopsied. Upper endoscopy showed a dysfunctional hiatus with chronic esophagitis. Biopsy of the neoplasm showed colorectal adenocarcinoma. The microsatellite technique was carried out in biopsy samples, and showed stability for all five microsatellites in the Bethesda panel (9), although the familial pedigree fulfils Amsterdam II criteria. Staging showed no metastatic spread.
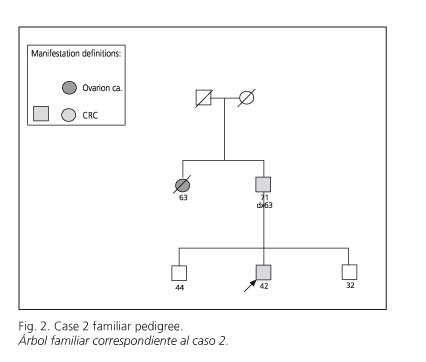
The patient underwent right hemicolectomy. A pathological study of the surgical specimen showed a mucinous, poorly differentiated, adenocarcinoma infiltrating perivisceral fat, with 21 lymph nodes free of disease (stage B2; pT3N0M0). Immunohistochemical testing for MMR proteins was carried out, which showed a normal expression of Mlh1, Msh2, and Msh6.
Case 3
A 28-year-old female with a family history of FAP was evaluated at the High Risk CRC Unit of our institution. She was asymptomatic, without any personal history of disease. Her family history is summarized in figure 3. A diagnostic colonoscopy was carried out in which more than 100 polyps were found along the whole length of the large bowel. Biopsies from some of them were reported as adenomatous polyps without dysplasia in any of them. She is the youngest affected member in the family, and that is the reason why a genetic analysis was carried out in her. A mutation in the APC gene was detected (exon 7, c.268 G > T) that generated a stop signal and a truncated protein. Due to these clinical and molecular findings, she is waiting for prophylactic surgery, in which a panproctocolectomy with ileal pouch-anal anastomosis will be carried out.

Discussion
Lynch syndrome
Lynch syndrome is the most common form of hereditary CRC, and is characterized by the development of CRC, endometrial cancer, and other malignancies at an early age. It is due to a germline mutation in one of the DNA mismatch repair genes (mismatch repair genes, MMR): MLH1, MSH2, MSH6 and PMS2. An alteration in one of these genes causes an accumulation of multiple errors in DNA repetitive sequences (microsatellites) along the genome. This finding is known as microsatellite instability (MSI), and is frequent but not exclusive in Lynch syndrome.
Surgical management
Once a CRC is diagnosed in a patient with Lynch syndrome a complete colonoscopy must be carried out in the first place, due to the high risk for a synchronous tumor. The elective surgical technique must take into account the risk of metachronous CRC during follow-up, which in some studies is as high as 16% at 10 years (10). For this reason subtotal colectomy with ileorectal anastomosis is recommended at the time CRC is diagnosed. According to the study by de Vos tot Nederveen Cappel WH et al. (11), if this operation is carried out at an early age (younger than 47 years) life expectancy is increased by 2.3 years. In this study data are not adjusted for quality of life, but the authors suggest that this should be increased as well, because the need of follow-up colonoscopies is reduced and so is concern about developing a new neoplasm. Our patient with Lynch syndrome (case 1) was in a follow-up program because of his family history of cancer. Although a genetic study was not performed before surgery for CRC, the possibility of Lynch syndrome could be assumed on the basis of absent Msh2 and Msh6 protein expression on immunohistochemistry testing, together with clinical suspicion (Amsterdam I family). Surgery was indicated on these grounds. It is controversial whether this type of surgery should be indicated without previous genetic testing. We think that, if the patient has given a fully informed consent, and with immunohistochemistry testing for MMR system proteins or MSI, it can be an acceptable indication, provided that the frequency of CRC in the family is high enough.
In healthy carriers of Lynch syndrome mutations some facts are to be considered when facing the possibility of indicating prophylactic surgery. The first one is that an effective and low-risk examination technique, namely colonoscopy, is widely available. Secondly, incomplete penetrance for CRC in this syndrome is as high as 70-80% (12). Lastly, according to recently published data, the risk of developing CRC decreases in older patients (13). For these reasons prophylactic colectomy should not be considered the operation of choice in this group of patients.
Prospective studies should be carried out to determine more precisely the cumulative risks at different ages, and on that basis to define the role of prophylactic surgery for healthy carriers who could benefit from it.
In the prevention of gynecological neoplasia some studies recommend that total hysterectomy with double anexectomy be carried out simultaneously with surgery for CRC (14) in patients with Lynch syndrome, but there are some families whose Lynch syndrome phenotype shows only CRC, and no other neoplasm. In these patients the indication for such operation is not as clear-cut, especially during their fertile period, as happened in our case.
Familial colorectal cancer type X syndrome
In the last few years a group of patients has been identified with a family tree that fulfils clinical criteria for Lynch syndrome, but that does not show the molecular features related to MMR system genes, as MSI and lack of expression of MMR proteins in the tumors (2-4). In this type X syndrome age at diagnosis of CRC is higher, CRC location is mainly in the left colon and rectum, and the proportion of mucinous tumors is smaller, and all features in contrast to Lynch syndrome. The family oncologic pedigree would be almost restricted to CRC. As a recently defined entity, there are no studies identifying its molecular basis, nor follow-up studies to assess the risk of metachronous CRC development, which seems to be smaller than in Lynch syndrome (3,15,16).
Surgical management
The lack of follow-up data in this group of patients underlines the fact that standard oncologic surgery is indicated instead of any kind of prophylactic operation. That is why, in case 2, although Amsterdam II criteria for Lynch syndrome were met, and particularly the fact that the patient showed an early-onset CRC, immunohistochemistry testing and the absence of MSI determined that standard, and not prophylactic surgery, was carried out.
Familial adenomatous polyposis
Familial adenomatous polyposis (FAP) is an autosomic dominant syndrome defined by the presence of more than a hundred colonic adenomatous polyps, mostly developing at a young age. The risk of malignant transformation is 100%. FAP represents less than 1% of all CRCs. Patients can develop polyps at any level of the digestive tract, with the duodenum being the most frequent location (17). Extraintestinal features, such as desmoid tumors, congenital hypertrophy of the pigmented retinal epithelium, thyroid neoplasia, hepatoblastoma, and brain tumors can also be found (18).
Surgical management
It is clear that carrying such a high risk of CRC development there is no discussing the need for prophylactic surgery in FAP (19), with some debate arising about which operation should be carried out. FAP patients without CRC found in any polyps, but in whom malignancy cannot be ruled out, must undergo prophylactic surgery. So it was indicated in our case (case 3), from the clinical, as well as the molecular and family history points of view.
The three available techniques are: panproctocolectomy with permanent ileostomy (PPCI); total colectomy (TC) with ileorectal anastomosis; and panproctocolectomy with ileoanal pouch (PPCR). The election of a particular technique depends on the desired quality of life after surgery and the need of follow-up for the risk of malignancy in the rectal remnant (19,20). PPCI is rarely the first option because of the consequences of permanent ileostomy, even if this is the technique with fewer surgical complications (21). Only technical considerations (short small bowel mesentery due to desmoid tumor) or sphincter muscle dysfunction determine the indication of this technique.
PPCR is preferred to TC due to a lesser risk of developing CRC in the rectal remnant, though this risk is not completely eliminated (23). CRC can develop not only in the pouch but in the anal transition area (24). The follow-up strategy depends on the operation performed. When a pouch is made endoscopy should be carried out every 2 to 5 years, while with TC the interval between explorations should be of 6 months. A temporary diverting ileostomy can be made to prevent anastomotic leakage when a pouch is used. Although PPCR is the preferred technique, controversy still exists regarding several technical aspects, such as transanal mucosectomy with sutured or stapled anastomosis, indications for laparoscopic access, a lesser than total mesorectal excision to prevent nerve damage, and in some cases to avoid temporary ileostomy (23).
Though it is clear that prophylactic surgery is the treatment of choice for FAP, there are certain circumstances in which delaying surgery can be considered, especially when there is a high risk of developing a desmoid tumor, which often grows on surgical scars. The use of chemopreventive agents such as COX-2 inhibitors should neither delay nor be substituted for surgical treatment.
Conclusions
As can be seen from the examples presented, especially in hereditary cases, the high risk of CRC, as in FAP, or of developing a metachronous tumor, does change surgical indications in this group of patients with CRC. For this reason it is important that the need to carry out tests as indicated in each case be underscored so that patients carrying a higher risk can be preoperatively identified in order to perform the best surgical technique. Nowadays, when clinical characteristics, associated cancer risks, and the molecular basis of family aggregation for CRC are unknown, surgical indications and techniques should remain as in standard cases, although an individualization of subjects and their families is mandatory. Essential criteria to be considered include age at diagnosis of CRC and family cancer history, as well as characteristic phenotypes, as in FAP. In the next few years, with the identification of the facts behind genetic susceptibility in CRC aggregations, prophylactic surgery will find a more important role than it currently has.
References
1. La situación del cáncer en España. Madrid: Ministerio de Sanidad y Consumo; 2005. [ Links ]
2. Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293: 1979-85. [ Links ]
3. Valle L, Perea J, Carbonell P, Fernández V, Dotor AM, Benítez J, et al. Clinicopathological and pedigree differences in Amsterdam I-positive hereditary nonpolyposis colorectal cancer families according to tumor microsatellite instability status. J Clin Oncol 2007; 25(7): 781-6. [ Links ]
4. Llor X, Pons E, Xicola RM, Castells A, Alenda C, Piñol V, et al. Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway. Clin Cancer Res 2005; 11: 7304-10. [ Links ]
5. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 2006; 101(2): 385-98. [ Links ]
6. Mecklin JP, Aarnio M, Läärä E, Kairaluoma MV, Pylvänäinen K, Peltomäki P, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133(4): 1093-8. [ Links ]
7. Vasen HF, Möslein G, Alonso A, Bernstein I, Bertario L, Blanco I, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44(6): 353-62. [ Links ]
8. You YN, Lakhani VT, Wells SA Jr. The role of prophylactic surgery in cancer prevention. World J Surg 2007; 31(3): 450-64. [ Links ]
9. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58(22): 5248-57. [ Links ]
10. De Vos tot Nederveen Cappel WH, Nagengast FM, Griffioen G, Menko FH, Taal BG, Kleibeuker JH, et al. Surveillance for hereditary nonpolyposis colorectal cancer: a long term study on 114 families. Dis Colon Rectum 2002; 45: 1588-94. [ Links ]
11. De Vos tot Nederveen Cappel WH, Buskens E, van Duijvendijk P et al. Decision analysis in the surgical treatment of colorectal cancer due to a mismatch repair gene defect. Gut 2003; 52: 1752-5. [ Links ]
12. Guillem JG, Smith AJ, Calle JP, Ruo. Gastrointestinal polyposis syndromes. Curr Probl Surg 1999; 36(4): 217-323. [ Links ]
13. Jenkins MA, Baglietto L, Dowty JG, Van Vliet CM, Smith L, Mead LJ, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006; 4(4): 489-98. [ Links ]
14. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296(12): 1507-17. [ Links ]
15. Jover R, Castells A. Molecular information defines a new entity of hereditary colorectal cancer. Gastroenterology 2008; 134(3): 888-9. [ Links ]
16. Valle L, Urioste M. Molecular information defines a new entity of hereditary colorectal cancer (Reply). Gastroenterology 2008; 134(3): 889-90. [ Links ]
17. Kashiwagi H, Spigelman AD. Gastroduodenal lesions in familial adenomatous polyposis. Surg Today 2000; 30(8): 675-82. [ Links ]
18. Herráiz M, Muñoz-Navas M. Recognition and management of hereditary colorectal cancer syndromes. Rev Es Enferm Dig 2009; 101(2): 125-32. [ Links ]
19. Cordero Fernández C, Pizarro Moreno A, Garzón Benavides M, García-Lozano R, Belda Laguna O, Sobrino S, et al. Follow-up alter surgical treatment of patients with familial adenomatous polyposis: results in a southern Spanish population. Rev Esp Enferm Dig 2007; 99(8): 440-5. [ Links ]
20. Bixquert M. Adherence to a postoperative follow-up protocol in patients with familial adenomatous polyposis may prevent colorectal cancer development. Rev Es Enferm Dig 2007; 99(8): 433-7. [ Links ]
21. Church J. In which patients do I perform IRA, and why? Fam Cancer 2006; 5: 237-40. [ Links ]
22. Al-Sukhni W, Aronson M, Gallinger S. Hereditary colorectal cancer syndromes: familial adenomatous polyposis and Lynch syndrome. In: Jatoi I, Martin RF, eds. Hereditary Cancer Syndromes. Surg Clin North Am 2008; 88(4): 819-42. [ Links ]
23. Kartheuser A, Stangherlin P, Brandt D, Remue C, Sempoux C. Restorative proctocolectomy and ileal pouch-anal anastomosis for familial adenomatous polyposis revisited. Fam Cancer 2006; 5(3): 241-60. [ Links ]
![]() Correspondence:
Correspondence:
José Perea García.
Servicio de Cirugía. Hospital 12 de Octubre.
Avda. de Córdoba, s/n. 28041 Madrid, Spain.
e-mail: josepereag@hotmail.com
Received: 28-01-09.
Accepted: 06-05-09.

