










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.102 no.4 Madrid abr. 2010
Barrett´s esophagus - a review
Esofago de Barrett
C. Ciriza-de-los-Ríos
Service of Digestive Diseases. Hospital Universitario "12 de Octubre". Madrid, Spain
Introduction and definition
Barrett´s esophagus (BE) is one of the most fascinating, challenging conditions in Gastroenterology. Its association with esophageal adenocarcinoma (ADC), explosive growth in Europe and the United States since 1980, and potential for easy diagnosis of preneoplastic lesions for follow-up and monitoring make it an appealing disease for both clinicians and researchers.
The condition was named after Dr. Norman Barrett, who in 1950 described an intrathoracic stomach secondary to "congenital short esophagus" (1). Shortly afterwards this mistake was rectified, and the condition was defined as a columnar metaplasia replacing the squamous epithelium at the distal esophagus (1), which clarified the fact that a short esophagus may be an equivocal observation, this being an esophagus that is normal in length but has a different inner epithelium. In 1961 Hayward suggested that only a metaplasia greater than 3 cm above the cardia was abnormal, as a transitional (cardial) mucosa between the esophageal squamous epithelium and the gastric columnar-oxyntic epithelium seemed a natural finding, and such transitional epithelium might be up to 3-cm long in any healthy adult. Interestingly, despite its purely speculative character, this description would become dogma for over 30 years (1). Chandrasoma et al. (2), in a necropsy study, revealed that this cardial epithelium (columnar mucosa bar parietal cells) was virtually absent in individuals younger than 20 years, and its presence and length increase with age, and suggested that this "transitional" epithelium was not normal but an acquired metaplasia. Oberg et al. (3) studied the association between cardial epithelium and acid exposure using distal esophageal pH-metry and esophageal manometry, and demonstrated that the cardial epithelium is significantly more common in subjects with gastroesophageal reflux (GER) and manometrically incompetent sphincter, thus concluding that this change may be caused by GER and is not attributable to infection with Helicobacter pylori, which is only found in 11% of subjects with cardial metaplasia.
These facts were determinant to our current understanding and definition of this disease (4). The condition is admittedly acquired, due almost exclusively to gastroesophageal reflux, but controversy remains when it comes to defining BE. The British and Japanese definition (5) consider that BE "is an esophagus where the squamous epithelium has been partly replaced by a metaplastic cylindrical epithelium that is macroscopically visible. A positive diagnosis with cylindrical metaplasia requires its observation above the gastroesophageal junction (GEJ) as well as histological confirmation". However, the definition in American and European guidelines consider BE the condition where intestinal metaplasia (IM) may be demonstrated by histology (6,7), and some authors state no BE may be diagnosed in the absence of goblet cells (8). The lack of a universally accepted definition for BE, with the aforementioned variants, has resulted in confusion and difficulties in comparing the various studies on this topic.
To make concepts clearer the British Gastroenterology Society has proposed the esophageal cylindrical epithelium concept, which would be a more descriptive term with the following categorization:
- Grade 0. No cylindrical epithelium or IM.
- Grade 1. Non-circumferential cylindrical epithelium, no IM.
- Grade 2. Non-circumferential cylindrical epithelium, with IM.
- Grade 3. Circumferential cylindrical epithelium, no IM.
- Grade 4. Circumferential cylindrical epithelium, with IM.
Anyway, to adequately diagnose BE, regardless of the definition used, the GEJ must be accurately identified, as well as the squamous-columnar junction where both epitheliums meet, the so-called Z line (9). GEJ is an imaginary line where the esophagus ends and the stomach begins anatomically. No anatomical structure truly delimiting the esophagus end and the start of the stomach exists. From an endoscopic viewpoint structures such as the vascular palisade or cardial narrowing have been reported, but there is now consensus that the best endoscopic description of GEJ is that related to the proximal border of gastric folds during partial insufflation (10). The hiatal imprint is also important to correctly identify hiatal hernia, a condition where the presence and length of columnar metaplasia may be more difficult to establish (11).
Pathology
A number of epitheliums may be found at a grossly normal GEJ according to necropsy studies: squamous, cardial, oxyntic, cardial oxyntic and intestinal (2). These epithelium types were previously described by Paull et al. (12). The definition of cardia is uncertain, as this term is used to define both the lower esophageal sphincter and the most proximal stomach. Cardial mucosa defines a type of epithelium with mucosal glands that may differentiate to parietal or intestinal cells. Previous studies defined a normal Z line as the junction between esophageal squamous epithelium and cardial epithelium. However, many authors currently consider that a normal Z line is the junction between squamous and fundic epithelium, and that cardial epithelium is abnormal, and consists of acquired metaplastic mucosa as a consequence of chronic inflammation at the distal esophagus from GER (13,14).
Previous studies such as the one by Csendes et al. (15) showed that the presence of cardial mucosa and carditis in patients with gastroesophageal reflux disease (GERD) may be a metaplasia of the fundic or squamous mucosa at the squamous-columnar junction, which is located within the lower esophageal sphincter (LES). This metaplasia may likely exclusively occur from early pathological acid reflux through the LES. As LES incompetence increases reflux becomes more massive towards the distal esophagus, and ascends towards its lining cardial mucosa. Another study with endoscopies on 959 patients with various indications found that the esophageal squamous mucosa merges with the gastric oxyntic mucosa in 19.9% of patients, whereas the remaining 80.1% have metaplastic mucosa between both epithelia, with 45.4% being cardial in type.
In this sense a new condition has been histologically defined and designated carditis; it results from chronic GER and consists of cardial mucosa proximal to the oxyntic (fundic) mucosa. The term "inflammation" needs not be included since the cardial mucosa is always inflamed. Biopsies may be labeled as "esophagus", "GEJ", "cardia", or "distal to GEJ". Histological findings consist of mucous cells and at times glands. It has chronic inflammation (eosinophils, plasma cells, lymphocytes in the lamina propria), and reactive changes (gland distortion, foveolar elongation, fibrosis, and smooth muscle proliferation at the lamina propria).
A columnar mucosa is visualized between the proximal border of gastric folds and squamous epithelium. A normal endoscopic exam may also ensue, and the condition is only detectable with biopsies immediately distal to the squamous epithelium. When severe it may have a polypoid appearance (16).
Some pathologists consider it a consequence of GERD so that in the absence of symptoms carditis defines "asymptomatic reflux". Patients usually have an abnormal pH-metry, and normal pH-metry would be a false negative result (16). Reflux carditis defines GERD -when present, a patient has cell-level GERD. When no carditis is demonstrated after adequate biopsy collection GERD may be excluded. Specificity is 100% and sensitivity is 100% (except for sampling errors) (16). A series of 141 patients with cardial mucosa in biopsies showed chronic inflammation and reactive changes even with a normal adjacent oxyntic mucosa. A correlation between inflammation severity and higher reflux severity in pH-metry was found. An inflammation of the proximal gastric mucosa or "gastric cardia" is no carditis but gastritis, and this may account for discrepancies in relating this condition to H. pylori infection (17). Hence some pathologists consider that when defined using stringent histological rather than anatomical criteria carditis is always secondary to GER. Regarding BE cardial, fundic, and intestinal epithelia may be found. A particularly interesting aspect of BE is a "mosaic" distribution of cell changes, usually with cardial metaplasia, intestinal metaplasia, and even areas of dysplasia. There will likely be more than just one metaplastic area, but successive stages with various metaplasias presumably occur (cardial-intestinal) (16). Different studies have shown that intestinal metaplasia is at the most proximal portion of the columnar epithelium (18). However, most important among clinical terms is the fact that only intestinal metaplasia may follow the sequence of low-grade dysplasia (LGD), high-grade dysplasia (HGD), and ADC (19). Hence most authors consider BE any columnar metaplasia endoscopically visible at the distal esophagus where histology demonstrates the presence of mucin-secreting goblet cells, which is characteristic of intestinal metaplasia (19).
However, recent studies showed that cardial mucosa is the most commonly found metaplasia in esophageal ADC (20), and that the presence of glandular mucosa with no intestinal metaplasia in the esophagus has a similar risk for neoplasia when compared to cases with intestinal metaplasia (21). These new data are leading to reconsider the need for goblet cells in esophageal biopsies for the diagnosis of BE. The problems entailed by this requirement for goblet cells in biopsies include the fact that these cells are rare in pediatric patients, a small proportion of adults with cylindrical metaplasia have no goblet cells, chances to demonstrate these cells are proportional to cylindrical metaplasia length, sampling errors are common, and differentiating goblet from pseudogoblet cells may be challenging. Metaplasia in BE with no goblet cells has been recently suggested to represent a number of molecular abnormalities similar to those in epithelia with goblet cells (22).
This controversy is important since a definition for BE has a number of clinical and economical implications. Therefore, diagnostic guidelines need to consider these new data on cylindrical epithelium with no goblet cells, and the challenging recognition of columnar metaplasia when < 1 cm (22).
As a premalignant lesion IM may progress to low-grade and high-grade intraepithelial dysplasia or neoplasia. LGD is characterized by preserved glandular architecture, and nuclei increased in both number and size that are usually elongated and stratified up to two thirds though never reaching the luminal third or pole of the cell, and never losing their perpendicular position regarding the basal membrane. There is nuclear hyperchromasia, presence of mitoses without atypical characteristics, and decreased cytoplasmic mucin. HGD presents with architectural distortion with gland branching, gemations, villous transformation at the mucosal surface, intraglandular epithelial bridges, compact gland clustering. A severe increase in the number and size of nuclei may be seen, which are pleomorphic, with irregular contour and nuclear hyperchromasia. There is also pronounced nuclear stratification with loss of nuclear polarity, and many nuclei reach the luminal pole. Mitoses increase in numbers, and atypical mitoses develop. Prominent nucleoli may be seen. There is usually no mucin secretion. It is considered an intraepithelial neoplasm given that the lamina propria is unscathed. ADC displays the above-mentioned changes plus complete loss of glandular architecture and lamina propria invasion (23).
The expression undefinite dysplasia is used when the presence or absence of dysplasia cannot be unequivocally established. Regenerative changes or acute inflammation are present. Cytological changes suggestive of deep tissue dysplasia (nuclear hyperchromasia, increased mitosis) may be found, however with superficial tissue maturity and preserved epithelial glandular architecture. Loss of nuclear polarity strongly suggests dysplasia. Most important is that this categorization leads to repeat biopsies after 2-3 months on intensive acid-suppressing therapy (23).
The estimated risk for IM progression to LGD, HGD, and ADC is 4.3%, 0.9%, and 0.5% per year, respectively. Progression from LGD to ADC: 0.6%/year; from HGD to ADC: 10%/year (24). Esophageal dysplasia is therefore currently considered the best risk marker for cancer in BE (25).
Another important aspect when planning potential therapy options is awareness of ADC infiltration extent in BE. A double muscularis mucosae (mm) has been found to be a histological characteristic of BE. Some pathologists may mistake a superficial mm for a single mm, and interpret infiltration beyond this first mm as submucosal invasion when the lesion has not truly reached the deep mm yet. In this sense Vieth et al. (26) have suggested a new classification of the various mucosal layers. When the tumor reaches the deep mm lymphatic spread may occur in 2.8-10% of cases (27). Pathology reports must accurately describe ADC infiltration extent rather than provide general descriptions, including mucosal or submucosal invasion.
Having histological aspects and columnar epithelium length in mind patients are categorized according to their risk for ADC development; patients with IM in biopsy samples would represent 5-15% of the population at-risk, whereas 55-65% of the population without cardiac or IM would have no ADC risk (16).
Pathophysiology and risk factors
GERD is the greatest risk factor associated with BE development (5,7). BE length has been suggested to correlate with the percentage of time with pH < 4 during both the total and supine stages of pH-metry recording (28). Other studies also show that reflux symptom duration, frequency, and severity are a risk factor for ADC development (29). While the prevalence of BE is higher in males, both males and females with BE share reflux severity, and the female gender does not protect from BE within the context of advanced GERD (30). The presence of BE is not associated with gastric acid hypersecretion since no differences in basal acid output or gastrin-stimulated peak acid output have been found by controlled studies (31). On the other hand, the nature of refluxed material is relevant, and bile reflux is increased in patients with BE when compared to controls and subjects with GERD and no BE (30,31).
The action of acid and pepsin weakens cell junctions and widens intracellular gaps, thus letting acid in. Acid and pepsin penetration allow acid to contact nerve endings. In turn, decreased pH favors conjugated bile acid deposition, which alters intracellular mechanisms and results in cell disrupture and damage. Mechanisms favoring this greater esophageal exposure to gastric contents and bile include LES hypotony (antireflux barrier changes) and hiatal hernia (HH) almost invariably in patients with BE; the latter is longer and associated with larger defects in the hiatus versus controls or patients with esophagitis with no BE (32). Other involved factors include esophageal motor abnormalities (esophageal clearance alterations), reduced esophageal sensitivity to pain, and decreased epidermoid growth factor secretion in the saliva (delayed esophageal healing) (19).
Patients with a long BE (LBE) have more commonly HH, greater esophageal exposure to acid, and more hypotense LES versus patients with a short BE (SBE) and intestinal metaplasia of the cardia (IMC) (33). GERD (reflux esophagitis and GER symptoms) are factors predictive for the development of angiogenesis in BE, which has malignant potential because epithelial cells express COX-2 and have accelerated cell proliferation (34,35).
Other factors such as obesity, which is an independent factor for BE and ADC development (36), have been involved even though two metaanalyses demonstrate no association between body mass index and BE in excess of that expected for GERD itself (37,38). Obesity may favor BE via GEJ disruption (lower LES pressure and HH) and an increase in GERD-favoring mechanisms (increased intraabdominal pressure and transient LES relaxations). However, studies increasingly explore the possibility that visceral fat may contribute to BE development via the production of factors such as leptin, adiponectin, and other cytokines. Leptin is of special interest because of its mitogenic and angiogenic effects (39,40). This cytokine has also been implicated in prostate cancer, breast cancer, and other gastrointestinal tumors (41).
Nitrogen compounds also play a role in the pathophysiology of BE. The saliva and diet are nitrogen sources as nitrates, which are reduced to nitrites by oral bacteria. These compounds are inert with neutral pH but become oxidative compounds with acid pH that may have mutagenic potential. The study by Suzuki (42) demonstrates that these potentially mutagenic nitrites act on the distal esophagus during reflux events. Conditions required to generate acid-catalyzed nitrogen compounds include pH < 4, nitrits in excess of ascorbic acid, and thiocyanate for catalyzer. During reflux episodes these conditions seemingly occur in the BE segment and to a lesser extent in hiatal hernia (42).
Helicobacter pylori does not seem to play a role in BE; Helicobacter pylori strains expressing cytokine associated to gene A (cagA) may even be a potential protective factor in decreasing acid production because of secondary gastritis (43,44). Studies on the pathogenetic role of tobacco (45) and alcohol yield conflictive results, and an association with BE is only found in some of them (46,47). Vegetable- and fruit-rich diets have been associated with a lower BE risk attributed to high antioxidant levels (48). Vitamin C plays an important role in reducing nitrogen compounds (42).
Therefore, when considering risk factors for BE, its development seems to require an esophageal mucosal lesion and a pathological environment allowing abnormal reepithelization (31). In either case, the reason why some patients with GERD have no lesions, other cases are complicated with esophagitis, and another set of patients develop BE remains unclear. BE metaplasia seems to develop rapidly and reach its maximum length in some studies, with few subsequent changes regarding length (49). The mechanism whereby damaging factors trigger metaplasia and why this occurs in a selected group of individuals remains unknown. The progenitor cell originating these changes is poorly established, and a most accepted hypothesis involves the differentiation of a pluripotential cell at the basal layer of the esophageal epithelium (50). Recent studies suggest that pluripotential bone marrow cells may contribute to esophageal lesion regeneration and metaplasia in BE (51). Despite all this, the reason why BE occurs in selected individuals is unclear, and genetic factors have been implied that may predispose to these changes. No specific gene has been found in this disease but the prevalence of GERD has been seen to be higher among relatives of affected persons and identical twins versus non-identical twins. Twin registers suggest that heritability accounted for 31 to 43% of GERD cases (52).
A higher familial aggregation than expected by chance has been confirmed for both BE and ADC. A recent study suggests that BE frequency among first-degree relatives of patients with BE with or without GER symptoms was 20 and 18%, respectively (53).
Advances in molecular biology have allowed the detection of abnormal expression in various genes that correlate with the transition from normal esophagus to ADC (54). Genetic changes may be found at a chromosomal or molecular level (Table I).
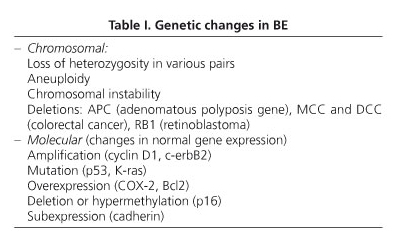
Epidemiology
The incidence of BE has increased from 1970 paralleling the increase in endoscopic exams (55). The study by Corley et al. (55) can check that the incidence has progressively increased both globally and adjusted to the number of endoscopies (55). This increase has been particularly obvious during the last few years. Mean age at diagnosis is usually around 50-55 years with a peak at 61-70 years, and rarely occurs before 5 years. The frequency of BE among the general population oscillates in the various studies between 0.9 and 4.5%. A Scandinavian study found a prevalence of 1.6% (56). By extrapolating these data the prevalence of BE in the USA is estimated around 3.3 million individuals (57). Many BE cases remain undetected in the general population, which renders the prevalence in autopsy studies higher than estimated in clinical studies (58,59).
BE predominates in males in a 2:1 proportion with respect to women in the Caucasian race (55).
The prevalence estimated in 961 patients undergoing colonoscopy for colon cancer screening was 6.8% (60). In this same study the prevalence found in patients with no GER symptoms was 5.6% (60) and 8.3% in patients with GERD symptoms (60). Most BE cases were short BE, long BE being extremely rare in patients with no GER symptoms (60). Therefore, BE may occur in subjects with no GERD symptoms. On the other hand the estimated prevalence of SBE is 10-15% and that of LBE is 3-5% (19). The prevalence of IMC is highly variable and oscillates around 6-36% (31).
The incidence of ADC has progressively increased whereas the incidence of squamous-cell carcinoma has declined. The estimated incidence in a meta-analysis including 47 studies was 6.1/1,000 person-years and 3.9/1000 person-years when only high-quality studies were included (61).
However, despite a higher frequency of ADC in patients with BE, only 4.7% died for that cause, and overall mortality in BE was similar to that of the general population (62). Another study found the BE increases the risk of death from ADC 20-fold, even though ADC is un uncommon cause of death in patients with BE (63).
Diagnosis
As suggested in the definition, regardless of which one is considered, a diagnosis with BE requires the identification of gastric metaplasia (cylindrical epithelium) in endoscopy, and its histological confirmation.
For an adequate endoscopic diagnosis there must be consensus when it comes to exploring the GEJ. There is no anatomical structure unequivocally separating the esophageal end from the beginning of the stomach. From an endoscopic point of view structures such as the vascular palisade or cardial narrowing have been described, but there is currently consensus that the best endoscopic description of GEJ is defined by the proximal limit of gastric folds during partial insufflation (10). The squamous-columnar junction or Z line macroscopically corresponds to an obvious, regular or irregular, circumferential colour change at the distal esophagus, which results from the border between the flat esophageal mucosa and the columnar gastric mucosa. The hiatal imprint corresponds to a distal narrowing of the lumen as seen during endoscopy. The term "hiatal" refers to an extrinsic origin resulting from the diaphragm crura.
However, in normal subjects at this level the presence of the LES must be considered. Csendes et al. (64), in 109 asymptomatic subjects, found a complete coincidence between the gastroesophageal sphincter midpoint and the squamous-columnar junction. In the presence of HH this constriction may be more sensibly designated "hiatal imprint". Studies using high-resolution esophageal manometry suggests that in patients with reflux, even in the absence of HH, there is separation between both sphincters (65). A normal gastroesophageal region is endoscopically defined by a coincidence in the three endoscopic items described: GEJ (proximal of gastric folds), squamous-columnar junction (Z line), and hiatal imprint. A difference of up to 1 cm between the Z line and GEJ is traditionally accepted as normality (even without endoscopic coincidence) to avoid overdiagnosis.
Despite emphasis on the importance of identifying the above-mentioned anatomical structures and measuring distance for the diagnosis of BE, lack of diagnostic consistency remains an issue. In a multicenter study anatomical structures were identified only by 71% of endoscopists, and pathological criteria for a diagnosis of BE were only followed by 70% of pathologists (66).
LBE is endoscopically defined when the distance from the Z line to GEJ is longer than 3 cm. Various studies have confirmed a higher incidence of ADC when BE is longer than 9 cm (5,67) and, therefore, that it is a risk factor for ADC. The SBE notion (distance from the Z line to GEJ lower than 3 cm) (68) is still a clinical dilemma regarding when and how biopsies should be obtained. However, SBE is also associated with a risk for dysplasia and ADC, and follow-up recommendations do not differ from those of LBE. Once esophageal metaplasia develops BE length does not considerably change over time (67).
It is important that the endoscopic diagnosis of BE be standardized. The Prague classification considers circumferential metaplasia (C) and maximum metaplasia extent (M), including tongues (11). This system leads to identify previously mentioned endoscopic marks (GEJ, Z line, hiatal imprint), extent of circumferential metaplasia, and proximal metaplasia tongues determining BE length. Reliability coefficients were 0.91 for C and 0.66 for M. In the external validation study 29 endoscopists scored 29 video tapes, and reliability coefficients obtained included 0.85 for the hiatus, 0.88 for the proximal border of gastric folds, 0.78 for distal esophageal compression, 0.94 for C, and 0.93 for M (11). This same analysis found an overall reliability coefficient for endoscopic recognition BE < 1 cm of 0.21, and of BE > 1 cm of 0.72. The primary conclusions to extract from this study include:
1. BE description including limits both nearest and farthest regarding GEJ is a reasonable approach to quantify extension.
2. The proximal limit of gastric folds is the most practical indicator of GEJ (minimal esophageal distension).
3. Mucosal changes suggesting a cylindrical epithelium < 1 cm above GEJ are of uncertain value for a diagnosis of BE.
4. Mucosal changes suggesting a cylindrical epithelium ≥ 1 cm above the GEJ probably predict BE.
Recent studies are demonstrating the usefulness of the Prague classification, increasing the prevalence of BE as a result of better recognition (69). Clinical guidelines consider that the endoscopic description of BE must be accurate and when feasible according to established classification systems (7,70).
Esophagitis may coexist with and at times mask up BE. In a study of 172 patients with esophagitis BE was diagnosed in 12% following esophagitis healing (71). It is therefore recommended that esophagitis be healed before biopsy collection in diagnosing BE (7). BE may also coexist with stenosis in 15-40% of cases in prospective studies, and with ulceration in 60%. Mucosal damage (stenosis and ulceration) is a risk factor for ADC (70).
A crucial aspect for BE confirmation is histology; hence biopsy collection standardization is a major issue. Different protocols have been suggested. The one by DeMeester requires biopsies from the stomach, GEJ, and every 1-2 cm of columnar metaplasia, all this processed and labeled as separate specimens (16). One of the better established systems is the Seattle protocol where biopsies must be collected from each esophageal quadrant at 1- or 2-cm intervals, and of visible changes such as nodules or ulcers, all as separate specimens (72). Regardless of the chosen protocol, biopsies should be collected from the most proximal columnar metaplastic area when diagnosing intestinal metaplasia (18).
The most adequate number of biopsies has been established as a minimum of eight as this allows a recognition of intestinal metaplasia in 68% of endoscopies versus 34.7% when biopsies number only four (73).
Anyway, no single protocol has been validated and established globally. The second European forum on endoscopy endorsed that jumbo forceps are not needed for biopsy collection, that biopsies are unwarranted for normal GEJ, and that biopsies from SBE tongues are recommended (74).
Standard endoscopy has limitations in the diagnosis and follow-up of BE since direct viewing cannot differentiate intestinal metaplasia from cardial mucosa, or assess dysplasia. Additionally, quadrant biopsy collection has moderate sensitivity, mainly as a result of a sampling error (75).
During the last few years multiple optic methods have been developed or refined to improve intestinal metaplasia and dysplasia detection. Chromoendoscopy is a simple technique involving the application of chemical staining agents. Vital staining with Methylene blue seems less sensitive than Seattle protocol to detect dysplasia (76). Three patterns have been described for chromoendoscopy with Indigo carmine: striated/villous (BE without dysplasia and with LGD), circular, and irregular/distorted (HGD), the latter with 100% sensitivity and specificity for HGD (77). Acetic acid seems better than conventional videoendoscopy with random biopsy collection for diagnosing BE (78 versus 57%, respectively) (78). Chromoendoscopy has a greater diagnostic yield when used together with high resolution endoscopy (up to 150x); it allows an assessment of specific patterns associated with dysplasia, and targeted biopsies with greater effectiveness for endoscopic surveillance (79). Despite such benefits the real usefulness of non-magnification chromoendoscopy and the lack of a consensus description of changes seen in endoscopic patterns are much debated topics, as well as the absence of controlled studies for techniques with magnification.
Some techniques are now available in clinical practice, but still no validated, that attempt to detect BE lesions and a better characterization of these based on dysplastic anatomic and functional changes. Narrow band imaging (NBI) has defined 3 mucosal patterns: striated/villous (BE without dysplasia and LGD), circular, and irregular/distorted (HGD); sensitivity and specificity are 100%, and 2 vascular patterns: normal and abnormal (characteristic of HGD) (80). Magnification or high resolution endoscopy (HRE) followed by NBI is equivalent to high resolution endoscopy followed by chromoendoscopy with indigo carmine (86 versus 93%, respectively) for HGD or early ADC detection in BE (81).
Other promising techniques include Fuji intelligent color chromoendoscopy (FICE), optical coherence tomography systems (OCT), and autofluorescence, with consensus that further validation and effectiveness assessment are needed before they can be implemented or recommended on a general basis.
In addition to endoscopist issues regarding an adequate diagnosis of BE, pathologists experience difficulties when defining BE from a histological viewpoint. The first issue is sampling error, which may be improved by using a well-defined protocol for biopsy collection like the Seattle protocol (72). Another issue is intraobserver and interobserver variability in that interobserver agreement for LGD is low, below 50%, whereas it reaches 85% for HGD (82).
In addition, active inflammation may induce nuclear changes simulating LGD and HGD. However, chronic inflammation does not induce such changes (82).
In view of histological issues other cytological techniques have been used including DNA analysis and fluorescence with in situ hibridation (FISH). The latter technique is highly sensitive in detecting HGD and ADC, and has the benefit of patient stratification for subjects with FISH changes by progression speed to HGD and ADC (83).
The presence of molecular markers (biomarkers) to select groups at risk of developing HGD or ADC, has increased the efficacy and cost-effectiveness of endoscopic surveillance. Flow cytometry (tetraploidy, aneuploidy) and p53 and p16 mutations (methylation, mutation, and loss of heterozygosity LOH) have been evaluated in prospective studies (84-87).
While results are promising further clinical research is needed to recommend its routine use (88).
Functional digestive tests share the same recommendations for BE and GERD. Manometry is indicated to verify the appropriate position of pH electrodes, and also allows an evaluation of antireflux barrier changes (LES hypotony, HH), and of the esophageal body motor activity (peristaltic dysfunction) (89).
Patients with BE often have no symptoms, and a way of checking the adequacy of proton-pump-inhibitors (PPIs) therapy is gastroesophageal pH-metry. In fact pH studies often demonstrate pathological acid reflux levels in patients with BE despite inhibitor therapy. In a study of patients with BE, GERD, and controls, 50% of patients with BE had abnormal esophageal acid exposure during nocturnal acid breakthrough (NAB) despite being asymptomatic (90). No predictive factors allow an identification of patients with BE and pathological esophageal acid exposure despite proton-pump inhibitors (PPIs). It has been suggested that these patients are resistant to therapy with PPIs, but such resistance results from their pathophysiological changes rather than failed gastric acid inhibition, so that the small amount of acid remaining in the stomach flows into the esophagus (91).
Bile reflux is a pathophysiological mechanism for BE development. Regarding its diagnosis, the presence of bile in the stomach is no evidence for pathological duodeno-gastro-esophageal reflux. The most accurate method for the study of bile reflux is the bilitec, a fiber optic device (a portable spectrometer with a 450-nm absorption peak, which represents bilirubin´s wavelength) (92). Operation is identical to pH-metry, that is, transnasally placing a sensor at 5 cm above the LES (92).
Treatment
Treatment of BE without dysplasia
Treatment of reflux
Medical
The goals of antireflux therapy include the control of symptoms management and the prevention of BE progression. However, many patients with BE have few or no symptoms because of columnar mucosal unresponsiveness to acid, hence controlled symptoms should not be interpreted as suppressed GER (70). Clinical management is similar to that of GERD patients without BE but is initiated with a PPI rather than less powerful antisecretory drugs. Therefore, PPIs are the drugs of choice in the treatment of reflux for patients with BE (89). In addition, all patients with BE, including asymptomatic subjects, should be treated for GERD. Patients with BE have higher acid exposure, and symptom management may require higher PPI doses (6). Adequate acid inhibition may be verified using pH-metry or bilitec. Should acid suppression be inadequate a prokinetic or anti-H2 agent may be added to prevent nocturnal acid breakthrough (93). A major issue with anti-H2 is the development of tachyphylaxis within the first week of therapy (93,94).
There is some debate on the need for aggressive antireflux therapy in all patients with BE regardless of reflux severity. Several longitudinal cohort studies have found that PPIs would reduce the risk for dysplasia in BE (95,96).
GERD is a well-known factor for ADC development, and significant acid suppression is deemed to potentially prevent tumor development. However, whether GERD facilitates ADC by causing an initial metaplasia (BE), by promoting carcinogenesis once BE is established, or both things remains unclear. In in vitro studies acid pulses promote cell proliferation in BE tissue (97), and reflux-related chronic inflammation might well promote carcinogenesis (97).
Surgical
Fundoplication has the advantage -at least theoretically- of correcting anatomical BE changes as LES hypotony, and HH, and of preventing acid and bile reflux (98). Laparoscopic antireflux surgery has proven effective and safe in the long-term management of BE (99). In some studies surgery seems to favor a less inflammatory and carcinogenetic environment versus medical therapy in patients with LBE (100). In other subjects it seemingly improves histological phenotype (extension and type of intestinal metaplasia) in patients with SBE but not LBE (101).
Fundoplication indications in BE are the same as in GERD; what happens is that patients with BE more commonly have HH, LES hypotony, and acid and bile reflux management difficulties, which renders indication more frequent in these patients versus other patients with GERD (89).
No conclusive studies exist regarding the possible preventive effect of surgery for ADC development. Fundoplication seems better than PPI therapy in observational studies regarding the incidence of ADC, but controlled studies find no differences (102).
A metaanalysis found no differences between fundoplication and medical treatment regarding the incidence of ADC, with a lower trend in patients undergoing surgery (103).
To conclude, the effect of surgery on the incidence of ADC is uncertain but there is seemingly a trend towards a reduced incidence. Therefore, surgery for ADC prevention cannot be currently recommended.
Treatment of metaplasia: edoscopic treatment
Endoscopic ablation
In the last few years endoscopic therapies have been proposed using thermal energy (argon plasma coagulation, laser, radiofrequency, multipolar electrocoagulation) or photochemical energy (PDT) to destroy BE. They all require intense acid suppression. Argon plasma coagulation (APC) requires 2-8 sessions and achieves a complete metaplastic remission in 32-100% of cases (104). Radiofrequency (HALO) leads to complete metaplastic remission (68-97%) with a mean follow-up of 3-30 months (105,106).
The role of this therapy for BE without dysplasia is not established as yet, and its use must be restricted to controlled research studies with this indication.
Treatment of BE with dysplasia and ADC
Esophagectomy, on eliminating the whole of dysplastic epithelium, has been the standard therapy for HGD and early ADC in patients with BE, with a 5-year survival probability greater than 90-95% (107). However, this technique entails significant morbidity, and a mortality rate of 3-5% in reference centers. Intramucosal ADCs have a very low risk for nodal involvement, which permits local management. Various endoscopic techniques have been developed to treat dysplasia and early ADC (Table II).
Endoscopic mucosal resection (EMR)
EMR, in contrast to ablation techniques, allows a histological assessment of lesions and defines both lateral infiltration margins and deep involvement (108). Conio et al. (109) and Mino-Kenudson et al. (110) reported a changed histological diagnosis in 26 and 37%, respectively, of patients with BE undergoing EMR. Studies suggest that EMR may successfully eliminate early ADC with a low complication rate but relapse rates of 25-30% during the first 3 years when used as single therapy in patients with HGD or intramucosal ADC (111). This fact represents one of this technique´s shortcomings since strict endoscopic follow-up or ablation of residual BE is required following EMR.
Circumferential mucosectomy is a step forward in resection that allows a complete, radical excision of metaplastic epithelium, offers optimal histological assessment, and prevents the persistence of residual BE spots. However, it is technically challenging and only indicated for BE segments with a length below 5 cm (112). It has a complication rate of 2% including hemorrhage and perforation, but the incidence of stenoses is high at 20-50% (107).
Radiofrequency (RDF)
Circumferential and/or focal BE ablation using RDF with a HALO system is a new promising technique. Initial circumferential ablation is performed with a balloon-coupled bipolar electrode whereas secondary treatment for residual BE areas is performed with a bipolar electrode fitted to the endoscope´s end. Recent data from clinical trials including patients with BE without dysplasia, with LGD or HGD, and with intramucosal ADC following EMR, demonstrate that RF ablation, combined or otherwise with EMR, is safe and effective for dysplasia and intestinal metaplasia eradication (113). Pouw et al. (114) treated with RDF 44 patients with BE plus LGD, HGD, or intramucosal ADC. EMR was performed in 31 cases before ablation. After 21 months of follow-up the authors described full dysplasia and intestinal metaplasia eradication in 98% of cases. A key point when comparing this technique with PDT is the absence of occult IM spots under the new squamous epithelium (115,116). Similarly, RDF ablation preserves esophageal function without inducing stenosis. Studies with a long follow-up are needed to confirm that BE clearance is sustained over time.
Chemoprevention
Observational studies show that NSAIDs reduce the risk for ADC by 50% (117). NSAID prophylaxis reduces biological markers (aneuploidy and tetraploidy) in BE (118). Most recent efforts have focused in COX-2 selective. Despite such encouraging evidence multicenter randomized studies (200 mg of celecoxib versus placebo) show no greater benefit (119). Extensive studies are being carried out to elucidate whether NSAIDs may play a role in ADC prevention in patients with BE.
While it was already noted that deep acid inhibition with a PPI may reduce the risk for dysplasia, PPIs are not indicated for ADC prevention. However, due to the presence of reflux and scarce side effects, acid inhibition is recommended in these patients as previously pointed out (120).
Population screening
Scientific societies are in conflict on this topic. Indications for endoscopy in patients with GERD symptoms are those recommended by the American Society for Endoscopy:
1. Presence of dysphagia or odinophagia.
2. Symptoms that persist or progress despite therapy.
3. Extraesophageal symptoms.
4. Esophageal symptoms in immunodepressed patients.
5. Alert symptoms: presence of a mass, stenosis or ulcers, digestive bleeding, iron-deficiency anemia or weight loss.
British guidelines consider that only patients with reflux and reflux-related alert symptoms should undergo endoscopy. Risk factors for cylindrical metaplasia development are well established: chronic GERD symptoms, male gender, obesity and age > 50 years. In this respect recommendations have been issued for endoscopy in patients older than 50 years (88) and long-standing reflux symptoms (121).
In a hypothetical screening model considering that absolute cancer counts near 8,000 yearly, and that many occur in subjects without significant reflux symptoms, over 10 million Americans would require screening even if only subjects older than 50 are considered. While endoscopy is a safe procedure, its associated risk would exceed the number of malignancies found (122).
General population screening is however controversial. Screening is not established for high-risk populations, hence it must be individualized (Table III).
Follow-up
BE without dysplasia
Patients with BE have a poorer quality of life as compared to the general population (123); in addition, they do not adequately understand and usually overestimate the frequency of malignancies associated with their disease (124). The risk for ADC in patients with BE is very low (0.5%/year) (125), and progression to ADC or HGD is below 10%. On the other hand mortality causes in patients with BE are similar to those in the general population. Some cost-effectiveness studies suggest a potential for more risk than benefit in follow-up programs (126). Therefore, there is some controversy when it comes to indicate such a program for patients with BE, even though most guidelines suggest some sort of follow-up.
Criteria to be met in order to include a patient in a follow-up program are as follows (7):
- Age.
- Survival chances for the next 5 years.
- Patient understanding about the process and limitations of ADC detection.
- Patient adherence to recommendations.
Taking these data into account most clinical guidelines recommend that patients with BE and no dysplasia should be treated conservatively with variations in patient follow-up length (5,7,88,121) (Table III and Fig. 1).
BE with dysplasia
When histology finds BE with dysplasia there is consensus in the various clinical guidelines that dysplasia should be confirmed by a second pathologist (5,7,25). In case of LGD several follow-up options are suggested in the different guidelines (5,7,88,121) (Table III and Fig. 1). There is greater controversy in the management of HGD given the risk of progression to ADC, and most consensus guidelines suggest esophagectomy, endoscopic ablation, or follow-up with biopsies every 3 months (5,7,88,121) (Table III and Fig. 1).
References
1. Burdiles P, Csendes A, Smok G, Braghetto I, Korn O. Progression from intestinal metaplasia to adenocarcinoma in Barrett´s esophagus: usefulness of endoscopic surveillance. Rev Med Chil 2003; 131: 587-96. [ Links ]
2. Chandrasoma PT, Der R, Ma Y, Dalton P, Taira M. Histology of the gastroesophageal junction: an autopsy study. Am J Surg Pathol 2000; 24: 402-9. [ Links ]
3. Oberg S, DeMeester TR, Peters JH, Hagen JA, Nigro JJ, DeMeester SR, et al. The extent of Barrett´s esophagus depends on the status of the lower esophageal sphincter and the degree of esophageal acid exposure. J Thorac Cardiovasc Surg 1999; 117: 572-80. [ Links ]
4. DeMeester SR, DeMeester TR. Columnar mucosa and intestinal metaplasia of the esophagus: fifty years of controversy. Ann Surg 2000; 231: 303-21. [ Links ]
5. Playford RJ. New British Society of Gastroenterology (BSG) guidelines for the diagnosis and management of Barrett's oesophagus. Gut 2006; 55: 442. [ Links ]
6. Sampliner RE. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett's esophagus. Am J Gastroenterol 2002; 97: 1888-95. [ Links ]
7. Wang KK, Sampliner RE. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett's esophagus. Am J Gastroenterol 2008; 103: 788-97. [ Links ]
8. Batts K. Barrett´s esophagus, more steps forward. Hum Pathol 2001; 32: 357-9. [ Links ]
9. Spechler SJ. Barrett's oesophagus: diagnosis and management. Baillieres Best Pract Res Clin Gastroenterol 2000; 14: 857-79. [ Links ]
10. Amano Y, Ishimura N, Furuta K, Takahashi Y, Chinuki D, Mishima Y, et al. Which landmark results in a more consistent diagnosis of Barrett's esophagus, the gastric folds or the palisade vessels? Gastrointest Endosc 2006; 64: 206-11. [ Links ]
11. Sharma P, Dent J, Armstrong D, Bergman JJ, Gossner L, Hoshihara Y, et al. The development and validation of an endoscopic grading system for Barrett's esophagus: the Prague C & M criteria. Gastroenterology 2006; 131: 1392-9. [ Links ]
12. Paull A, Trier JS, Dalton MD, Camp RC, Loeb P, Goyal RK. The histologic spectrum of Barrett's esophagus. N Engl J Med 1976; 295: 476-80. [ Links ]
13. Chandrasoma P. Pathophysiology of Barrett's esophagus. Semin Thorac Cardiovasc Surg 1997; 9: 270-8. [ Links ]
14. Dresner SM, Griffin SM, Wayman J, Bennett MK, Hayes N, Raimes SA. Human model of duodenogastro-oesophageal reflux in the development of Barrett's metaplasia. Br J Surg 2003; 90: 1120-8. [ Links ]
15. Csendes A, Smok G, Christensen H, Rojas J, Burdiles P, Korn O. Prevalencia de mucosa cardial o fúndica y presencia de Helicobacter pylori en la unión de mucosas escamoso-columnar en pacientes con reflujo gastroesofágico crónico patológico sin metaplasia intestinal comparados con controles. Rev Med Chile 1999; 127: 1439-46. [ Links ]
16. Chandrasoma P. Controversies of the cardiac mucosa and Barrett's oesophagus. Histopathology 2005; 46: 361-73. [ Links ]
17. Goldblum JR, Richter JE, Vaezi M, Falk GW, Rice TW, Peek RM. Helicobacter pylori infection, not gastroesophageal reflux, is the major cause of inflammation and intestinal metaplasia of gastric cardiac mucosa. Am J Gastroenterol 2002; 97: 302-11. [ Links ]
18. Chandrasoma PT, Der R, Dalton P, Kobayashi G, Ma Y, Peters J, et al. Distribution and significance of epithelial types in columnar-lined esophagus. Am J Surg Pathol 2001; 25: 1188-93. [ Links ]
19. Spechler SJ. Clinical practice. Barrett's esophagus. N Engl J Med 2002; 346: 836-42. [ Links ]
20. Takubo K, Aida J, Naomoto Y, Sawabe M, Arai T, Shiraishi H, et al. Cardiac rather than intestinal-type background in endoscopic resection specimens of minute Barrett adenocarcinoma. Hum Pathol 2009; 40: 65-74. [ Links ]
21. Kelty CJ, Gough MD, Van Wyk Q, Stephenson TJ, Ackroyd R. Barrett's oesophagus: intestinal metaplasia is not essential for cancer risk. Scand J Gastroenterol 2007; 42: 1271-4. [ Links ]
22. Riddell RH, Odze RD. Definition of Barrett's esophagus: time for a rethink-is intestinal metaplasia dead? Am J Gastroenterol 2009; 104: 2588-94. [ Links ]
23. Guindi M, Riddell RH. Histology of Barrett's esophagus and dysplasia. Gastrointest Endosc Clin N Am 2003; 13: 349-68, viii. [ Links ]
24. Sharma P, Falk GW, Weston AP, Reker D, Johnston M, Sampliner RE. Dysplasia and cancer in a large multicenter cohort of patients with Barrett's esophagus. Clin Gastroenterol Hepatol 2006; 4: 566-72. [ Links ]
25. Wang KK, Wongkeesong M, Buttar NS. American Gastroenterological Association technical review on the role of the gastroenterologist in the management of esophageal carcinoma. Gastroenterology 2005; 128: 1471-505. [ Links ]
26. Vieth M, Ell C, Gossner L, May A, Stolte M. Histological analysis of endoscopic resection specimens from 326 patients with Barrett's esophagus and early neoplasia. Endoscopy 2004; 36: 776-81. [ Links ]
27. Lewis JT, Wang KK, Abraham SC. Muscularis mucosae duplication and the musculo-fibrousanomaly in endoscopic mucosal resections for Barrett esophagus: implications for staging of adenocarcinoma. Am J Surg Pathol 2008 32: 566-71. [ Links ]
28. Fass R, Hell RW, Garewal HS, Martinez P, Pulliam G, Wendel C, et al. Correlation of oesophageal acid exposure with Barrett's oesophagus length. Gut 2001; 48: 310-3. [ Links ]
29. Lagergren J, Bergström R, Lindgren A, Nyrren O. Symptomatic gastroesophageal refux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999; 340: 825-31. [ Links ]
30. Banki F, Demeester SR, Mason RJ, Campos G, Hagen JA, Peters JH, et al. Barrett's esophagus in females: a comparative analysis of risk factors in females and males. Am J Gastroenterol 2005; 100: 560-7. [ Links ]
31. Falk GW. Barrett's esophagus. Gastroenterology 2002; 122: 1569-91. [ Links ]
32. Cameron AJ. Barrett's esophagus: prevalence and size of hiatal hernia. Am J Gastroenterol 1999; 94: 2054-9. [ Links ]
33. Csendes A, Smok G, Quiroz J, Burdiles P, Rojas J, Castro C, et al. Clinical, endoscopic, and functional studies in 408 patients with Barrett's esophagus, compared to 174 cases of intestinal metaplasia of the cardia. Am J Gastroenterol 2002; 97: 554-60. [ Links ]
34. Moriyama N, Amano Y, Mishima Y, Okita K, Takahashi Y, Yuki T, et al. What is the clinical significance of stromal angiogenesis in Barrett's esophagus? J Gastroenterol Hepatol 2008; 23(Supl. 2): S210-5. [ Links ]
35. Shirvani VN, Ouatu-Lascar R, Kaur BS, Omary MB, Triadafilopoulos G. Cyclooxygenase 2 expression in Barrett's esophagus and adenocarcinoma: Ex vivo induction by bile salts and acid exposure. Gastroenterology 2000; 118: 487-96. [ Links ]
36. Kubo A, Corley DA. Body mass index and adenocarcinomas of the esophagus or gastric cardia: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 872-8 . [ Links ]
37. Cook MB, Wild CP, Everett SM, Hardie LJ, Bani-Hani KE, Martin IG, et al. Risk of mortality and cancer incidence in Barrett's esophagus. Cancer Epidemiol Biomarkers Prev 2007; 16: 2090-6. [ Links ]
38. Cook MB, Greenwood DC, Hardie LJ, Wild CP, Forman D. A systematic review and meta-analysis of the risk of increasing adiposity on Barrett's esophagus. Am J Gastroenterol 2008; 103: 292-300. [ Links ]
39. Kendall BJ, Macdonald GA, Hayward NK, Prins JB, Brown I, Walker N, et al. Leptin and the risk of Barrett's oesophagus. Gut 2008; 57: 448-54. [ Links ]
40. Edelstein ZR, Farrow DC, Bronner MP, Rosen SN, Vaughan TL. Central adiposity and risk of Barrett's esophagus. Gastroenterology 2007; 133: 403-11. [ Links ]
41. Bird-Lieberman EL, Fitzgerald RC. Barrett's esophagus. Gastroenterol Clin North Am 2008; 37: 921-42, x. [ Links ]
42. Suzuki H, Iijima K, Scobie G, Fyfe V, McColl KE. Nitrate and nitrosative chemistry within Barrett's oesophagus during acid reflux. Gut 2005; 54: 1527-35. [ Links ]
43. Vaezi MF, Falk GW, Peek RM, Vicari JJ, Goldblum JR, Perez-Perez GI, et al. CagA-positive strains of Helicobacter pylori may protect against Barrett's esophagus. Am J Gastroenterol 2000; 95: 2206-11. [ Links ]
44. Anderson LA, Murphy SJ, Johnston BT, Watson RG, Ferguson HR, Bamford KB, et al. Relationship between Helicobacter pylori infection and gastric atrophy and the stages of the oesophageal inflammation, metaplasia, adenocarcinoma sequence: results from the FINBAR case-control study. Gut 2008; 57: 734-9. [ Links ]
45. Kubo A, Levin TR, Block G, Rumore G, Quesenberry CP, Jr., Buffler P, et al. Cigarette smoking and the risk of Barrett's esophagus. Cancer Causes Control 2009; 20: 303-11. [ Links ]
46. Anderson LA, Cantwell MM, Watson RG, Johnston BT, Murphy SJ, Ferguson HR, et al. The association between alcohol and reflux esophagitis, Barrett's esophagus, and esophageal adenocarcinoma. Gastroenterology 2009; 136: 799-805. [ Links ]
47. Kubo A, Levin TR, Block G, Rumore GJ, Quesenberry CP, Jr., Buffler P, et al. Alcohol types and sociodemographic characteristics as risk factors for Barrett's esophagus. Gastroenterology 2009; 136: 806-15. [ Links ]
48. Kubo A, Levin TR, Block G, Rumore GJ, Quesenberry CP, Jr., Buffler P, et al. Dietary antioxidants, fruits, and vegetables and the risk of Barrett's esophagus. Am J Gastroenterol 2008; 103: 1614-23. [ Links ]
49. Cameron AJ, Lomboy CT. Barrett's esophagus: age, prevalence, and extent of columnar epithelium. Gastroenterology 1992; 103: 1241-5. [ Links ]
50. Glickman JN, Chen YY, Wang HH, Antonioli DA, Odze RD. Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett's esophagus. Am J Surg Pathol 2001; 25: 569-78. [ Links ]
51. Sarosi G, Brown G, Jaiswal K, Feagins LA, Lee E, Crook TW, et al. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett's esophagus. Dis Esophagus 2008; 21: 43-50. [ Links ]
52. Cameron AJ, Lagergren J, Henriksson C, Nyren O, Lock GR III, Pedersen NL. Gastroesophageal reflux disease in monozygotic and dizygotic twins. Gastroenterology 2002; 122: 55-9. [ Links ]
53. Conio M, Blanchi S, Lacchin T, De Ceglie A, De Matthaeis M, Grossi E, et al. Endoscopy in first degree relatives of Barrett's esophagus patients. Gastroenterology 2009; 136: A-453. [ Links ]
54. Dahlberg PS, Ferrin LF, Grindle SM, Nelson CM, Hoang CD, Jacobson B. Gene expression profiles in esophageal adenocarcinoma. Ann Thorac Surg 2004; 77: 1008-15. [ Links ]
55. Corley DA, Kubo A, Levin TR, Block G, Habel L, Rumore G, et al. Race, ethnicity, sex and temporal differences in Barrett's oesophagus diagnosis: a large community-based study, 1994-2006. Gut 2009; 58: 182-8. [ Links ]
56. Ronkainen J, Aro P, Storskrubb T, Johansson SE, Lind T, Bolling-Sternevald E, et al. Prevalence of Barrett's esophagus in the general population: an endoscopic study. Gastroenterology 2005; 129: 1825-31. [ Links ]
57. Sampliner RE. A population prevalence of Barrett's esophagus--finally. Gastroenterology 2005; 129: 2101-3. [ Links ]
58. Cameron AJ, Zinsmeister AR, Ballard DJ, Carney JA. Prevalence of columnar-lined (Barrett's) esophagus. Comparison of population-based clinical and autopsy findings. Gastroenterology 1990; 99: 918-22. [ Links ]
59. Ormsby AH, Kilgore SP, Goldblum JR, Richter JE, Rice TW, Gramlich TL. The location and frequency of intestinal metaplasia at the esophagogastric junction in 223 consecutive autopsies: implications for patient treatment and preventive strategies in Barrett's esophagus. Mod Pathol 2000; 13: 614-20. [ Links ]
60. Rex DK, Cummings OW, Shaw M, Cumings MD, Wong RK, Vasudeva RS, et al. Screening for Barrett's esophagus in colonoscopy patients with and without heartburn. Gastroenterology 2003; 125: 1670-7. [ Links ]
61. Yousef F, Cardwell C, Cantwell MM, Galway K, Johnston BT, Murray L. The incidence of esophageal cancer and high-grade dysplasia in Barrett's esophagus: a systematic review and meta-analysis. Am J Epidemiol 2008; 168: 237-49. [ Links ]
62. Anderson LA, Murray LJ, Murphy SJ, Fitzpatrick DA, Johnston BT, Watson RG, et al. Mortality in Barrett´s oesophagus: results from a population based study. Gut 2003; 52: 1081-4. [ Links ]
63. Moayyedi P, Burch N, Akhtar-Danesh N, Enaganti SK, Harrison R, Talley NJ, et al. Mortality rates in patients with Barrett´s oesophagus. Aliment Pharmacol Ther 2008; 27: 316-20. [ Links ]
64. Csendes A, Maluenda F, Braghetto I, Csendes P, Henriquez A, Quesada MS. Location of the lower oesophageal sphincter and the squamous columnar mucosal junction in 109 healthy controls and 778 patients with different degrees of endoscopic oesophagitis. Gut 1993; 34: 21-7. [ Links ]
65. Pandolfino JE, Kim H, Gohsh SK, Clarke JO, Hang Q, Kahrilas PJ. High-resolution manometry of the EGJ: an analysis of crural diaphragm function in GERD. Am J Gastroenterol 2007; 102: 1056-63. [ Links ]
66. Ofman JJ, Shaheen NJ, Desai AA, Moody B, Bozymski EM, Weinstein WM. The quality of care in Barrett's esophagus: endoscopist and pathologist practices. Am J Gastroenterol 2001; 96: 876-81. [ Links ]
67. Gatenby PA, Ramus JR, Caygill CP, Watson A. Does the length of the columnar-lined esophagus change with time? Dis Esophagus 2007; 20: 497-503. [ Links ]
68. Sharma P, Morales TG, Sampliner RE. Short segment Barrett's esophagus--the need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol 1998; 93: 1033-6. [ Links ]
69. Chang CY, Lee YC, Lee CT, Tu CH, Hwang JC, Chiang H, et al. The application of Prague C and M criteria in the diagnosis of Barrett's esophagus in an ethnic Chinese population. Am J Gastroenterol 2009; 104: 13-20. [ Links ]
70. Murphy SJ, Johnston BT, Murray LJ. British Society of Gastroenterology guidelines for the diagnosis of Barrett's oesophagus: are we casting the net too wide? Gut 2006; 55: 1821-2. [ Links ]
71. Hanna S, Rastogi A, Weston AP, Totta F, Schmitz R, Mathur S, et al. Detection of Barrett's esophagus after endoscopic healing of erosive esophagitis. Am J Gastroenterol 2006; 101: 1416-20. [ Links ]
72. Levine DS, Haggitt RC, Blount PL, Rabinovitch PS, Rusch VW, Reid BJ. An endoscopic biopsy protocol can differentiate high-grade dysplasia from early adenocarcinoma in Barrett's esophagus. Gastroenterology 1993; 105: 40-50. [ Links ]
73. Wani S, Sharma P. The rationale for screening and surveillance of Barrett's metaplasia. Best Pract Res Clin Gastroenterol 2006; 20: 829-42. [ Links ]
74. Axon A, Lambert R, Robaszkiewicz M, Rosch T, Sonnenberg A. The Second European Endoscopy Forum. Twenty questions on the esophagogastric junction. Endoscopy 2000; 32: 411-8. [ Links ]
75. Padda S, Ramirez FC. Accuracy in the diagnosis of short-segment Barrett's esophagus: the role of endoscopic experience. Gastrointest Endosc 2001; 54: 605-8. [ Links ]
76. Lim CH, Rotimi O, Dexter SP, Axon AT. Randomized crossover study that used methylene blue or random 4-quadrant biopsy for the diagnosis of dysplasia in Barrett's esophagus. Gastrointest Endosc 2006; 64: 195-9. [ Links ]
77. Sharma P, Weston AP, Topalovski M, Cherian R, Bhattacharyya A, Sampliner RE. Magnification chromoendoscopy for the detection of intestinal metaplasia and dysplasia in Barrett's oesophagus. Gut 2003; 52: 24-7. [ Links ]
78. Hoffman A, Kiesslich R, Bender A, Neurath MF, Nafe B, Herrmann G, et al. Acetic acid-guided biopsies after magnifying endoscopy compared with random biopsies in the detection of Barrett's esophagus: a prospective randomized trial with crossover design. Gastrointest Endosc 2006; 64: 1-8. [ Links ]
79. Conio M. Esophageal chromoendoscopy in Barrett's esophagus: "cons". Gastrointest Endosc 2006; 64: 9-12. [ Links ]
80. Sharma P, Bansal A, Mathur S, Wani S, Cherian R, McGregor D, et al. The utility of a novel narrow band imaging endoscopy system in patients with Barrett's esophagus. Gastrointest Endosc 2006; 64: 167-75. [ Links ]
81. Kara MA, Peters FP, Rosmolen WD, Krishnadath KK, ten Kate FJ, Fockens P, et al. High-resolution endoscopy plus chromoendoscopy or narrow-band imaging in Barrett's esophagus: a prospective randomized crossover study. Endoscopy 2005; 37: 929-36. [ Links ]
82. Montgomery E, Bronner M, Goldblum J, Greenson J, Haber M, Hart J, et al. Diagnostic reproducibility of dysplasia in Barrett esophagus (BE): are affirmation. Hum Pathol 2001; 32: 368-78. [ Links ]
83. Fritcher EG, Brankley SM, Kipp BR, Voss JS, Campion MB, Morrison LE, et al. A comparison of conventional cytology, DNA ploidy analysis, and fluorescence in situ hybridization for the detection of dysplasia and adenocarcinoma in patients with Barrett's esophagus. Hum Pathol 2008; 39: 1128-35. [ Links ]
84. Reid BJ, Prevo LJ, Galipeau PC, Sanchez CA, Longton G, Levine DS, et al. Predictors of progression in Barrett's esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol 2001; 96: 2839-48. [ Links ]
85. Reid BJ, Levine DS, Longton G, Blount PL, Rabinovitch PS. Predictors of progression to cancer in Barrett's esophagus: baseline histology and flow cytometry identify low- and high-risk patient subsets. Am J Gastroenterol 2000; 95: 1669-76. [ Links ]
86. Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett's esophagus III: Baseline flow cytometric variable. Am J Gastroenterol 2001; 96: 3071-83. [ Links ]
87. Illueca C, Llombart-Bosch A, Ferrando Cucarella J. Prognostic factors in Barrett's esophagus: an immunohistochemical and morphometric study of 120 cases. Rev Esp Enferm Dig 2000; 92: 726-37. [ Links ]
88. Sharma P, McQuaid K, Dent J, Fennerty MB, Sampliner R, Spechler S, et al. A critical review of the diagnosis and management of Barrett's esophagus: the AGA Chicago Workshop. Gastroenterology 2004; 127: 310-30. [ Links ]
89. Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101: 1900-20; quiz 1943. [ Links ]
90. Katz PO, Anderson C, Khoury R, Castell DO. Gastro-oesophageal reflux associated with nocturnal gastric acid breakthrough on proton pump inhibitors. Aliment Pharmacol Ther 1998; 12: 1231-4. [ Links ]
91. Spechler SJ, Sharma P, Traxler B, Levine D, Falk GW. Gastric and esophageal pH in patients with Barrett's esophagus treated with three esomeprazole dosages: a randomized, double-blind, crossover trial. Am J Gastroenterol 2006; 101: 1964-71. [ Links ]
92. Hak NG, Mostafa M, Salah T, El-Hemaly M, Haleem M, Abd El-Raouf A, et al. Acid and bile reflux in erosive reflux disease, non-erosive reflux disease and Barrett's esophagus. Hepatogastroenterology 2008; 55: 442-7. [ Links ]
93. Tutuian R, Castell DO. Nocturnal acid breakthrough - approach to management. MedGenMed 2004; 6: 11. [ Links ]
94. Tutuian R, Katz PO, Castell DO. Nocturnal acid breakthrough: pH, drugs and bugs. Eur J Gastroenterol Hepatol 2004; 16: 441-3. [ Links ]
95. Hillman LC, Chiragakis L, Shadbolt B, Kaye GL, Clarke AC. Proton-pump inhibitor therapy and the development of dysplasia in patients with Barrett's oesophagus. Med J Aust 2004; 180: 387-91. [ Links ]
96. El Serag HB, Aguirre T, Davis S, Kuebeler M, Bhattacharyya A, Sampliner E. Proton pump inhibitors are associated with reduced incidence of dysplasia in arrett´s esophagus. Am J Gastroenterol 2004; 99: 1877-83. [ Links ]
97. Kaur BS, Triadafilopoulos G. Acid- and bile-induced PGE(2) release and hyperproliferation in Barrett's esophagus are COX-2 and PKC-epsilon dependent. Am J Physiol Gastrointest Liver Physiol 2002; 283: G327-34. [ Links ]
98. Ochando Cerdan F, Hernandez Garcia-Gallardo D, Moreno Gonzalez E. Barrett's esophagus control after antireflux surgery. Rev Esp Enferm Dig 2002; 94: 188-200. [ Links ]
99. Biertho L, Dallemagne B, Dewandre JM, Jehaes C, Markiewicz S, Monami B, et al. Laparoscopic treatment of Barrett's esophagus: long-term results. Surg Endosc 2007; 21: 11-5. [ Links ]
100. Babar M, Ennis D, Abdel-Latif M, Byrne PJ, Ravi N, Reynolds JV. Differential molecular changes in patients with asymptomatic long-segment Barrett's esophagus treated by antireflux surgery or medical therapy. Am J Surg 2010; 199: 137-43. [ Links ]
101. Zaninotto G, Cassaro M, Pennelli G, Battaglia G, Farinati F, Ceolin M, et al. Barrett's epithelium after antireflux surgery. J Gastrointest Surg 2005; 9: 1253-60; discussion 1260-1. [ Links ]
102. Chang EY, Morris CD, Seltman AK, O'Rourke RW, Chan BJ, Hunter JG, et al. The effect of antireflux surgery on esophageal carcinogenesis in patients with Barrett esophagus. A systematic review. Ann Surg 2007; 246: 11-21. [ Links ]
103. Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett's esophagus? A meta-analysis. Am J Gastroenterol 2003; 98: 2390-4. [ Links ]
104. Sharma P, Wani S, Weston AP, Bansal A, Hall M, Mathur S, et al. A randomised controlled trial of ablation of Barrett´s oesophagus with multipolar electrocoagulation versus argon plasma coagulation in combination with acid suppression: long term results. Gut 2006; 55: 1233-9. [ Links ]
105. Sharma VK, Wang KK, Overholt BF, Lightdale CJ, Fennerty MB, Dean PJ, et al. Balloon-based, circumferential, endoscopic radiofrequency ablation of Barrett´s esophagus: 1-year follow-up of 100 patients. Gastrointest Endosc 2007; 65: 185-95. [ Links ]
106. Fleischer DE, Overholt BF, Sharma VK, Reymunde A, Kimmey MB, Chuttani R, et al. Endoscopic ablation of Barrett's esophagus: a multicenter study with 2.5-year follow-up. Gastrointest Endosc 2008; 68: 867-76. [ Links ]
107. Pera M, Grande L, Iglesias M, Ramon JM, Conio M. New advances in the diagnosis and treatment of early onset dysplasia and adenocarcinoma in Barrett's oesophagus. Cir Esp 2009; 85: 331-40. [ Links ]
108. Espinel J, Pinedo, E, Rascarachi G. Endoscopic mucosal resection with a multiband ligator for the treatment of Barrett´s high-grade dysplasia and early gastric cancer. Rev Esp Enferm Dig 2009; 101: 403-7. [ Links ]
109. Conio M, Repici A, Cestari R, Blanchi S, Lapertosa G, Missale G, et al. Endoscopic mucosal resection for high-grade dysplasia and intramucosal carcinoma in Barrett's esophagus: an Italian experience. World J Gastroenterol 2005; 11: 6650-5. [ Links ]
110. Mino-Kenudson M, Hull MJ, Brown I, Muzikansky A, Srivastava A, Glickman J, et al. EMR for Barrett's esophagus-related superficial neoplasms offers better diagnostic reproducibility than mucosal biopsy. Gastrointest Endosc 2007; 66: 660-6; quiz 767, 769. [ Links ]
111. May A, Gossner L, Pech O, Fritz A, Gunter E, Mayer G, et al. Local endoscopic therapy for intraepithelial high-grade neoplasia and early adenocarcinoma in Barrett's oesophagus: acute-phase and intermediate results of a new treatment approach. Eur J Gastroenterol Hepatol 2002; 14: 1085-91. [ Links ]
112. Soehendra N, Seewald S, Groth S, Omar S, Seitz U, Zhong Y, et al. Use of modified multiband ligator facilitates circumferential EMR in Barrett's esophagus (with video). Gastrointest Endosc 2006; 63: 847-52. [ Links ]
113. Ganz RA, Overholt BF, Sharma VK, Fleischer DE, Shaheen NJ, Lightdale CJ, et al. Circumferential ablation of Barrett's esophagus that contains high-grade dysplasia: a U.S. Multicenter Registry. Gastrointest Endosc 2008; 68: 35-40. [ Links ]
114. Pouw RE, Wirths K, Eisendrath P, Sondermeijer CM, Ten Kate FJ, Fockens P, et al. Efficacy of radiofrequency ablation combined with endoscopic resection for Barrett's esophagus with early neoplasia. Clin Gastroenterol Hepatol 2010; 8: 23-9. [ Links ]
115. Pouw RE, Gondrie JJ, Rygiel AM, Sondermeijer CM, ten Kate FJ, Odze RD, et al. Properties of the neosquamous epithelium after radiofrequency ablation of Barrett's esophagus containing neoplasia. Am J Gastroenterol 2009; 104: 1366-73. [ Links ]
116. Sharma VK, Jae Kim H, Das A, Wells CD, Nguyen CC, Fleischer DE. Circumferential and focal ablation of Barrett's esophagus containing dysplasia. Am J Gastroenterol 2009; 104: 310-7. [ Links ]
117. Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology 2003; 124: 47-56. [ Links ]
118. Vaughan TL, Dong LM, Blount PL, Ayub K, Odze RD, Sanchez CA, et al. Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett's oesophagus: a prospective study. Lancet Oncol 2005; 6: 945-52. [ Links ]
119. Heath EI, Canto MI, Piantadosi S, Montgomery E, Weinstein WM, Herman JG, et al. Secondary chemoprevention of Barrett's esophagus with celecoxib: results of a randomized trial. J Natl Cancer Inst 2007; 99: 545-57. [ Links ]
120. Shaheen NJ. Advances in Barrett's esophagus and esophageal adenocarcinoma. Gastroenterology 2005; 128: 1554-66. [ Links ]
121. Standards of Practice Committee of the American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of endoscopy in the surveillance of premalignant conditions of the upper GI tract. Gastrointest Endosc 2006; 63: 570-80. [ Links ]
122. Shaheen N, Ransohoff DF. Gastroesophageal reflux, Barrett esophagus, and esophageal cancer: scientific review. JAMA 2002; 287: 1972-81. [ Links ]
123. Eloubeidi MA, Provenzale D. Health-related quality of life and severity of symptoms in patients with Barrett's esophagus and gastroesophageal reflux disease patients without Barrett's esophagus. Am J Gastroenterol 2000; 95: 1881-7. [ Links ]
124. Shaheen NJ, Green B, Medapalli RK, Mitchell KL, Wei JT, Schmitz SM, et al. The perception of cancer risk in patients with prevalent Barrett's esophagus enrolled in an endoscopic surveillance program. Gastroenterology 2005; 129: 429-36. [ Links ]
125. Shaheen NJ, Richter JE. Barrett's oesophagus. Lancet 2009; 373: 850-61. [ Links ]
126. Garside R, Pitt M, Somerville M, Stein K, Price A, Gilbert N. Surveillance of Barrett's oesophagus: exploring the uncertainty through systematic review, expert workshop and economic modelling. Health Technol Assess 2006; 10: 1-142. [ Links ]
![]() Correspondence:
Correspondence:
Constanza Ciriza de los Ríos.
Servicio de Aparato Digestivo.
Hospital Universitario 12 de Octubre.
Ctra. de Andalucía km. 5,400.
28041 Madrid, Spain.
Received: 23-11-09.
Accepted: 24-11-09.

