










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.104 no.3 Madrid mar. 2012
https://dx.doi.org/10.4321/S1130-01082012000300002
p16 gene methylation in colorectal cancer patients with long-term follow-up
Metilación de p16 en pacientes intervenidos de cáncer colorrectal tras un largo periodo de seguimiento
Silvia Veganzones-de-Castro1, Sara Rafael-Fernández1, Marta Vidaurreta-Lázaro1, Virginia de-la-Orden1, Beatriz Mediero-Valeros1, Cristina Fernández2 y María Luisa Maestro-de las Casas1
1Department of Clinical Analysis. Hospital Clínico San Carlos. Madrid, Spain.
2Research Unit. Hospital Clínico San Carlos. Madrid, Spain
ABSTRACT
Introduction: p16 gene plays an important role in the cell cycle regulation and is considered an important tumor suppressor gene. Several mechanisms of gene inactivation have been described; in this study we have focused on p16 gene promoter methylation. In colorectal cancer p16 gene methylation is a frequent event.
Methods: 326 patients with sporadic colorectal cancer were included. DNA was extracted from tumor tissue samples obtained during the surgical procedure. Promoter methylation was analyzed using bisulfite modification and was detected by quantitative methylation-specific PCR. Frequency of p16 methylation was analyzed and compared with other clinicopathological variables.
Results: p16 gene methylation was detected in 24,8% of patients. Methylation was associated with differentiation grade and with tumor location: methylation was frequent in poorly differentiated tumors and had low frequency in distal colon. The p16 promoter methylation discriminated a subgroup of patients with better prognosis in poorly differentiated tumors.
Conclusions: p16 methylation was a frequent event in our population and was able to induce differences in the overall survival of patients with poorly differentiated tumors.
Key words: p16 gene. Methylation. Colorectal cancer. Prognosis.
RESUMEN
Introducción: el gen p16 está implicado en la regulación del ciclo celular y se considera un importante gen supresor de tumores.
Objetivos: se han descrito diferentes mecanismos de inactivación génica, en este estudio nos hemos centrado en la metilación del promotor del gen p16. En el cáncer colorrectal la metilación de p16 es una alteración frecuente.
Material y métodos: se incluyeron 326 pacientes con cáncer colorrectal esporádico. El ADN se extrajo de muestras tumorales obtenidas durante la cirugía. La metilación del promotor se analizó mediante un proceso de modificación con bisulfito y posterior PCR cuantitativa especifica para metilación. Se analizó la frecuencia de la metilación de p16 y se comparó con las variables clinicopatológicas.
Resultados: la metilación del gen p16 se detectó en el 24,8% de los pacientes. La metilación de p16 se relacionó con el grado de diferenciación y con la localización tumoral: la metilación fue mas frecuente en los tumores pobremente diferenciados y tuvo una baja frecuencia en el colon distal. La metilación del promotor de p16 discrimina un subgrupo de pacientes con mejor pronóstico en los tumores pobremente diferenciados.
Conclusiones: la metilación de p16 es un evento frecuente en nuestra población y es capaz de inducir diferencias en la supervivencia global de los pacientes con tumores moderadamente diferenciados.
Palabras clave: Gen p16. Mutilación. Cáncer colorrectal. Pronóstico.
Introduction
Great efforts have been done to improve prediction of prognosis in colorectal patients (1-3). They have been focused on the genetic mechanisms implicated in tumorogenesis of colorectal cancer. The p16INK4a gene acts as a tumor suppressor gene and has associated with tumorogenesis when it is inactivated (4). This inactivation often occurs through promoter methylation. It is still unclear the prognosis influence of p16 in colorectal patients.
The p16 gene is also known as CDKN2, MTS1, INK4a and CDK4I. It is implicated in the cell cycle control, playing an important role as tumor suppressor gene (4). This gene is located on region 9p21, comprised of 3 exons, and codes for a 16 kDa protein. p16 inhibits the cyclin kinase D1-CDK4/6 complex, responsible for the phosphorylation of protein Rb, causing cell cycle arrest at G1 stage (5).
Alterations in the 9p21 gene region are frequent in human cancers (6). Several mechanisms for p16 gene inactivation have been described: deletion, promoter methylation and point mutation, but their incidence depends on tumor type (7). Point mutations in p16 are a rare event in human cancer and one of the most frequent causes for gene inactivation is loss of heterozygosity in 9p21 (8).
The CpG dinucleotides located in promoter region are called CpG islands. These regions are target for methylation as an important mechanism of transcriptional regulation (9,10). Methylation of promoter regions plays an important role in silencing of tumor suppressor genes and other genes in tumorogenesis. De novo methylation of CpG sequences in p16, CDH1, MGMT, and APC genes promoter has been described in approximately 30% of colorectal tumors (11-14).
The aim of our study was to determine the prevalence and to analyze the prognostic relevance of p16 promoter methylation using quantitative methylation-specific PCR (qMSP) in a wide cohort of patients with colorectal cancer with long follow-up.
Patients and methods
Sample collection and DNA preparation
The study cohort comprised 326 patients undergoing surgery consecutively for colorectal cancer at the Hospital Clínico San Carlos in Madrid (Spain) between 1995 and 2003. This is a prospective cohort study. All the patients were operated on by the same surgeon who performed radical oncological surgery, based on the location of the tumor. The surgery was defined as curative when there was no evidence of macroscopic residual tumor after resection. Using this criterion, the surgeon performed a curative resection in 269 patients (82.5%) and resected the primary tumor in 57 patients (17.5%) as palliative treatment. Patients with metacronic carcinoma, familial polyposis, familial predisposition for hereditary nonpolyposis colon cancer and inflammatory bowel disease were excluded from the study. None of the patients had received neoadjuvant treatment. Informed consent was obtained from each patient and the project was approved by the clinical research and ethics committee of this hospital. Follow-up was performed according to the protocol designed by the authors (15). Tumors were staged according to Duke's classification. Proximal tumors were defined as occurring in the cecum through to the transverse colon; tumors in the splenic flexure, descending and sigmoid colon were defined as being distal. Stage B and C patients received adjuvant treatment with 5-fluoracil and leucovorin (75% of the patients included). For stage D patients, different protocols were applied according to the Oncology Service criteria.
Tumor and non-tumor tissue samples were obtained during the surgical procedure and immediately immersed in liquid nitrogen for storage in a freezer at -80 oC. The specimens were then independently examined by two pathologists, who confirmed the samples had more than 80% tumor cells.
For the analysis, DNA was extracted from tumor tissue samples using DNeasy® Blood & Tissue Kit (Qiagen) following the manufacture's instructions.
qMSP
One µg of genomic DNA was subjected to bisulfite modification treatment using and Epitect Bisulfite Kit (Qiagen, Hilden, Germany). The bisulfite-treated DNA was amplified by qMSP, conducted in a Thermal Cycler Real-time System (Smart Cycler, CEPHEID). DNA integrity and modification reaction proficiency were verified using myogenic differentiation 1 (MYOD) gene as internal reference gene. A CpG island free region was selected because its amplification is independent of methylation status. The ratio between Ct value of target gene and Ct value of internal reference gene (MYOD) was used as a measure of the relative methylation level (RML = Ct p16/Ct MYOD), as described previously (16-18). Amplification was carried out in a final volume of 25 ml with QuantiTect Probe PCR Kit (Qiagen, Hilden, Germany) using 1 ml of bisulfite-modified DNA, 10 pmol of each primer and 5 pmol of probes. Primers and probes used in this reaction were:5'-TGGAGTTTTCGGTTGATTGGTT-3'(p16 sense), 5'-TCCTCCACGCCCGCAACAA-3' (p16 antisense), 5'-CGCCAAGCCCCAGCCCA-3'(p16 probe), 5'-GGATTTATATTTATGTGGTGGGTGG-3' (MYOD sense), 5'-TATCTCTCCCCTAAACCTCAACC-3'(MYOD antisense) and 5'-TAGGGGATAGAGGGAGGTGTTTAGGTTG-3' (MYOD probe). Primers and probes chosen for p16 amplification contained 7 CpG islands and 9 points for bisulfite modification detection (9 cytosines not included in a CpG island). CpG islands discriminate gene methylation status. Negative control (leukocytes from healthy men) and positive control (CpGenomeTM Universal Methylated DNA [Chemicon]) were included in each reaction (Fig. 1). The RML cut-off value for this study was fixed at 1.0 for p16 methylation. In thirty patients with proven methylation the non-tumor sample was analysed and none of them had p16 methylation.

Statistical analysis
Qualitative variables are provided with their corresponding frequency distributions. Quantitative variables are expressed as their mean, standard deviation and range. Associations between qualitative variables were evaluated using the χ2 test or Fisher's exact test when 25% of expected frequencies fell below 5. Overall survival (OS) and disease-free survival (DFS) were estimated by the Kaplan-Meier method and compared among groups using Breslow's exact test. The event in OS was defined as death occurring as a consequence of tumor, censuring live patients and those dying of another cause. OS was calculated as the time elapsed between the date of surgery the date of death or last follow-up. The event in DFS was defined as a diagnosis of locoregional or distant recurrence in patients free from disease. Patients undergoing palliative surgery were excluded for the DFS analysis. The data were fitted to Cox's proportional risks regression model. In each contrast, the null hypothesis was rejected when the type I error was equal or less than 0.05. All statistical tests were performed using SSPS v.11.5 software.
Results
In 326 patients recruited, 53.4% were men and 46.6% women and the median age was 71 years (range 35-95 years). Age was stratified according to median value. Clinicopathological variables are shown in table 1. 62.9% of the tumors were located in colon and 37.1% in rectum. 8.6% of tumors were mucinous adenocarcinomas. In 34 patients differentiation grade could not be assigned.
p16 methylation could not be analyzed in 8 patients. Methylation was positive in 24.8% of samples (79 of 318). In the analysis of p16 and the clinicopathological variables (Table I), we found statistically significant association of methylation and differentiation grade (p = 0.04). In undifferentiated tumors the frequency of methylation was higher (69.2%) than in moderately or well differentiated tumors (24.6 and 21.7%, respectively). Association of p16 methylation with tumor location was also found (p = 0.02). Methylation was lower in distal colon (16%) than in proximal colon or in rectum (32.4 and 25.9%, respectively). No other significant relation was observed in this analysis.

Postoperative course. Overall survival
Median follow-up period was 92 months; with an interquartile range from 75 to 111 months. In our population the OS of our population at this time was 63%. All survival analyses were referred to the median follow-up. During follow-up 142 patients died, 106 of them as a consequence of their neoplasia, and one patient was lost to follow-up. Results of the univariate analysis of OS are shown in table II.
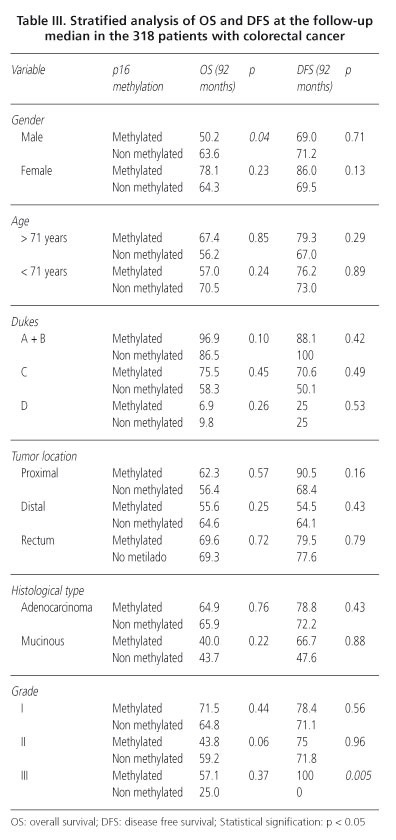
No differences in OS were observed according to p16 methylation status. OS after 92 months in patients showing p16 methylation was 62.6 and 64% in the patients without methylation -Hazard ratio-(HR) = 0.91; 95% confidence interval (95% CI = 0.58-1.42). A stratified analysis of OS was performed using the clinicopathological variables. According to this analysis, significant differences were observed in sex (Table III). In males with p16 methylation the OS was shorter than males without (50.2 vs. 63.6%; p = 0,04). We also found a tendency towards negative effect of p16 in OS of patients with moderately differentiated tumors, OS was 43.8% in those with methylation compared to 59.2% in patients without gene methylation (p = 0.06) (Fig. 2). In multivariable analysis we did not find statistically significant results for p16 methylation, Dukes stage was the only independent prognostic factor. We performed an exploratory analysis and the corrected HR for the absence of methylation of p16 was 0.88 (95% CI = 0.56-1.39).

Postoperative course. Disease-free survival
DFS in the median follow-up time was 71.6%. Tumor recurrence occurred in 69 patients. The recurrence was locoregional in 14 patients (20%) and distant in 49 patients (80%). Results of the DFS univariate analysis are shown in table II.
In patients with p16 methylation DFS was 78.2% and in those without this alteration was 70.3% (p = 0.3). Stratified analysis of DFS was performed according to the clinicopathological variables (Table III). Only in differentiation grade a significant effect was proved. Undifferentiated tumors with p16 methylation 0% showed recurrence while in absence of methylation 100% relapsed (p = 0.005) (Fig. 3). In the multivariable analysis for DFS the effect of p16 methylation was not independent prognostic factor.

Discussion
CRC is the second leading cause of death related to cancer in USA and Europe (19). It is one of the cancers with better characterised tumorogenesis genetic pathways and one of the common alterations implicated in tumorogenesis is DNA methylation (20). Promoter methylation silences tumor suppressor genes in great number of neoplasias (21,22). p16 gene acts as a negative controller of cell cycle playing an important role as tumor suppressor (23).
None of the previously published studies of p16 showed high population size, with so extended follow-up in homogeneous population, only with sporadic cases with no previous treatment (24-29). The only cases who reported higher population size with survival analyses in long follow-up whether had lower median follow up (23) whether consisted of multicenter studies, introducing variability through diverse laboratory analyses and different clinical management, including family history cases (25). In this cohort of 326 patients with sporadic CRC, we evaluated the frequency of p16 promoter methylation. These frequencies, for CRC patients, have been previously shown to be highly variable, ranging from 18 to 61% (30-36). This variability could be explained by the used of different methodologies in methylation analysis. We previously tested p16 methylation with methylation specific conventional PCR finding 18.3% positive results (33). However, the technique (qMSP) used by the current study increased the positive percentage to 24.8%. qMSP technique can both provide quantitative data, and greater sensitivity; even with small amounts of DNA it is able to detect up to 0.1% methylated alleles in a known CpG island (37).
Methylation of p16 gene promoter, for CRC patients, is a frequent event. The relation of p16 methylation with clinicopathological variables, in these patients, has yield discrepancies in the literature. Although methylation could not be associated to clinicopathological variables by some authors (28,38), it has been associated to the following: Dukes stage and lymphatic invasion (35); also to age, sex, tumor location, differentiation grade and histological type (36). In this study, the p16 gene promoter methylation was found to be associated with both, poor differentiation grade and proximal tumor location. Increased frequency of p16 methylation in proximal and poor differentiated tumors has been previously described by Shima et al. (25). Some authors have described previously described the increased p16 methylation detected in MSI-H tumors, which are preferentially located in proximal colon (39). MSI status has not been included in our study but could be influencing differences in p16 tumor location. Relation of p16 loss of expression, often caused by promoter methylation, and poor differentiation has been described in other malignancies (40,41). Absence of p16 expression causes deregulation in cell cycle and may lead to rapid cellular division and favours dedifferentiation process.
Overall methylation and also p16 methylation has been associated with age but this relation only appeared in our cohort as a statistical tendency (25,42). Other authors showed no association of p16 with age, this would explain why none of the normal mucosa analysed showed methylation (43).
Some studies have analysed survival and p16 promoter methylation (25,26,28,29). In prognosis studies, to work on homogeneous populations and with a long follow-up increases the value of the information obtained (median follow-up 92 months). Although methylation could not be associated to prognosis by some authors, it has been associated with a shorter overall survival (25,28). No significant effect of p16 methylation in prognosis was observed. However, in the preliminary multivariate analysis, a negative effect on OS was detected, but not de-monstrated, due to the population size. Shima et al. demonstrated in a multivariate analysis the significantly decrease in OS associated with the presence of methylation in p16 (25).
According to the p16 methylation status, no differences were found in the disease free survival. Some authors have found shorter DFS in the patients showing methylation (29). Unpredictably, in the poor differentiation group (Grade III) we found protective effect of p16 methylation: none of the patients showing methylation had recurrence. Other authors had not analysed the effect of p16 in this specific subgroup. The positive effect of p16 methylation in the prognosis has also been described in gastric tumors (29).
p16 methylation is frequent and induces differences in the DFS of patients with poorly differentiated tumors. However further studies increasing our population size are needed to elucidate p16 effect on the prognosis.
References
1. Vidaurreta M, Maestro ML, Rafael S, Veganzones S, Sanz-Casla MT, Cerdán J, Arroyo M. Telomerase activity in colorectal cancer, prognostic factor and implications in the microsatellite instability pathway. World J Gastroenterol 2007 Jul 28;13(28):3868-72. [ Links ]
2. Maestro ML, Vidaurreta M, Sanz-Casla MT, Rafael S, Veganzones S, Martínez A, Aguilera C, Herranz MD, Cerdán J, Arroyo M. Role of the BRAF mutations in the microsatellite instability genetic pathway in sporadic colorectal cancer. Ann Surg Oncol 2007;14(3):1229-36. [ Links ]
3. Vidaurreta M, Sánchez-Muñoz R, Veganzones S, Rafael S, Gutiérrez M, de-la-Orden V, Fernández C, Arroyo M, Cerdán FJ, Maestro de las Casas ML. Vascular endothelial growth factor gene polymorphisms in patients with colorectal cancer. Rev Esp Enferm Dig 2010;102(1):20-31. [ Links ]
4. Liggett WH and Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol 1998;16:1197-206. [ Links ]
5. Serrano M, Hannon GJ and Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993;366:704-7. [ Links ]
6. Nobori P, Miura K, Wu DJ, Lois A, Takabayashi K and Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994;368:753-6. [ Links ]
7. Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995;55:4525-30. [ Links ]
8. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS-1 in human cancers. Nature Med 1995;1:686-692. [ Links ]
9. Bird A. The essentials of DNA methylation. Cell 1992;70:5-8. [ Links ]
10. Perea J, Lomas M, Hidalgo M. Bases moleculares del cáncer colorrectal: ¿Hacia un manejo individualizado? Rev Esp Enferm Dig 2011;103:29-35. [ Links ]
11. Esteller M, Corn PG, Baylin SB and Herman JG. A gene hypermethylation profile of human cancer. Cancer Res 2001;61:3225-9. [ Links ]
12. Lind GE, Thorstensen L, Løvig T, Meling GI, Hamelin R, Rognum TO, et al. A CpG island hypermethylation profile of primary colorectal carcinomas and colon cancer cells lines. Mol Cancer 2004;3:28. [ Links ]
13. Lee S, Hwang KS, Lee HJ, Kim JS and Kang GH. Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab Invest 2004;84:884-93. [ Links ]
14. Noda H, Mashima R, Kamiyama H, Okada S, Kawamura YJ and Konishi F. Promoter hypermethylation of tumor-related genes in sporadic colorectal cancer in young patients. J Exp Clin Cancer Res 2007;26: 251-6. [ Links ]
15. Cerdán J. Recidiva locorregional en el cáncer de recto. Cir Esp 2003; 73:63-7. [ Links ]
16. Topaloglu O, Hoque MO, Tokumaru Y, Lee J, Ratovitski E, Sidransky D and Moon CS. Detection of promoter hypermethylation of multiple genes in the tumor and bronchoalveolar lavage of patients with lung cancer. Clin Cancer Res 2004;10:2284-8. [ Links ]
17. Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH, et al. Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine and serum DNA of patients with renal cancer. Cancer Res 2004;64:5511-7. [ Links ]
18. Carvalho AL, Jeronimo C, Kim MM, Henrique R, Zhang Z, Hoque MO, et al. Evaluation of promoter hypermethylation detection in body fluids as a screenning/diagnosis tool for head and neck squamous cell carcinoma. Clin Cancer Res 2008;14:97-107. [ Links ]
19. Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol 2001;2:533-43. [ Links ]
20. Jones PA and Laird PV. Cancer epigenetics comes of age. Nat Genet 1999;21:163-7. [ Links ]
21. Jones PA and Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002;3:415-28. [ Links ]
22. Noda H, Miyaji Y, Nakanishi A, Konishi F and Miki Y. Frequent reduced expression of alpha-1B-adrenergic receptor caused by aberrant promoter methylation in gastric cancers. Br J Cancer 2007;96:383-90. [ Links ]
23. Sharpless NE. INK4a/ARF A multifunctional tumor supresor locus. Mutat Res 2005;576:22-38. [ Links ]
24. Norrie MW, Hawkins NJ, Todd AV, Meagher AP, O'Connor TW and Ward RL. Inactivation of p16INK4a by CpG hypermethylation is not a frequent event in colorectal cancer. J Surg Oncol 2003;84:143-50. [ Links ]
25. Shima K, Nosho K, Baba Y, Cantor M, Meyerhardt JA, Giovannucci EL, et al. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expresión in 902 colorectal cancers: Cohort study and literatura review. Int J Cancer 2011;128:1080-94. [ Links ]
26. Ward RL, Cheong K, Ku SL, Meagher A, O'Connor T and Hawkins NJ. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J Clin Oncol 2003;21:3729-3736. [ Links ]
27. Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS ONE 2008;3:e3698. [ Links ]
28. Kamiyama H, Noda H, Takata O, Suzuki K, Kawamura Y and Konishi F. Promoter hypermethylation of tumor related genes in peritoneal lavage and the prognosis of patients with colorectal cancer. Journal of Surgical Oncology 2009;100:69-74. [ Links ]
29. Mitomi H, Fukui N, Tanaka N, Kanazawa H, Saito T, Matsuoka T, Yao T, et al. Aberrant p16INK4a methylation is a frequent event in colorectal cancers: prognostic value and relation to mRNA expresion and immunoreactivity. J Cancer Clin Oncol 2010;136:323-331. [ Links ]
30. Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB and Herman JG. Hypermethylation-associated inactivation of p14ARF is independent of p16INK4a methylation and p53 mutational status. Cancer Res 2000;60:129-33. [ Links ]
31. Guan RJ, Fu Y, Holt PR and Pardee AB: Asocciation of k-ras with p16 methylation of the p16 gene in human colon cancer. Gastroenterology 1999;116:1063-71. [ Links ]
32. Kanai Y, Ushijima S, Kondo Y, Nakanishi Y and Hirohashi S. DNA methyltransferase expression and DNA methylation of CpG islands and peri-centromeric satellite regions in human colorectal and stomach cancer. In J Cancer 2001;91:2005-212. [ Links ]
33. Sanz-Casla MT, Maestro ML, Vidaurreta M, Maestro C, Arroyo M and Cerdán J. p16 gene methylation in colorectal tumors: correlation with clinicopathological features and prognostic value. Dig Dis 2005;23:151-5. [ Links ]
34. Winencke JK, Zheng S, Lafuente A, Lafuente MJ, Grudzen C, Wrensch MR et al. Aberrant methylation of p16INK4a in anatomic and gender-spcific subtypes of sporadic colorectal cancer. Cancer Epidemiol Biomark Prev 1999;8:501-6. [ Links ]
35. Ogino S, Odze RD, Kawasaki T, Brahmandam M, Kirkner GJ, Laird PW et al. Correlation of pathologic features with CpG island methylator phenotype (CIMP) by quantitative DNA methylation analysis in colorectal carcinoma. Am J Surg Pathol 2006;30:1175-83. [ Links ]
36. Iacopetta B, Grieu F, Li W, Ruszkiewicz A, Caruso M, Moore J, et al. APC gene methylation is inversely correlated with features of the CpG island methylator phenotype in colorectal cancer. Int J Cancer 2006;119:2272-8. [ Links ]
37. Herman JG, Graff JR, Myöhänen S, Nelkin BD and Baylin SB. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996;93:9821-6. [ Links ]
38. Brücher B, Geddert H, Langner C, Höfler H, Fink U, Siewert JR and Sarbia M. Hypermethylation of hMLH1, HPP1, p14ARF, p16INK4A and APC in primary adenocarcinomas of the small bowel. In J Cancer 2006;119:1298-302. [ Links ]
39. Ahuja N, Mohan AL, Li Q, Stolker JM, Herman JG, Hamilton SR, Baylin SB, Issa JP. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res 1997;57:3370-4. [ Links ]
40. Kawabuchi B, Moriyama S, Hironaka M, Fujii T, Koike M, Moriyama H, Nishimura Y, Mizuno S, Fukayama M. p16 inactivation in small-sized lung adenocarcinoma: its association with poor prognosis. Int J Cancer 1999;84:49-53. [ Links ]
41. Tsujie M, Yamamoto H, Tomita N, Sugita Y, Ohue M, Sakita I, Tamaki Y, Sekimoto M, Doki Y, Inoue M, Matsuura N, Monden T, Shiozaki H, Monden M. Expression of tumor suppressor gene p16(INK4) products in primary gastric cancer. Oncology 2000;58(2):126-36. [ Links ]
42. Holliday R. The inheritance of epigenetic defects. Science 1987;238: 163-70. [ Links ]
43. Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998;58:5489-94. [ Links ]
![]() Correspondence:
Correspondence:
Ma Luisa Maestro de las Casas.
Deptartment of Clinical Analysis.
Hospital Clínico San Carlos.
C/ Martín Lagos s/n.
28040 Madrid, Spain.
e-mail: mmaestro.hcsc@salud.madrid.org
Received: 31-05-11.
Accepted: 21-10-11.

