










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.105 n.3 Madrid Mar. 2013
https://dx.doi.org/10.4321/S1130-01082013000300010
Collagenous sprue: Don't forget connective tissue in chronic diarrhea evaluation
Esprúe colágeno: no olvidemos el tejido conectivo en la evaluación de la diarrea crónica
Victoria Busto-Bea1, Laura Crespo-Pérez1, Noemí García-Miralles2, Luis Ruiz-del-Árbol-Olmos1 and Ana Cano-Ruiz1
Departments of 1Gastroenterology and 2Pathology. Hospital Universitario Ramón y Cajal. Madrid, Spain
ABSTRACT
Collagenous sprue is a rare disease of the small bowel characterized by mucosal atrophy and excessive subepithelial collagen deposition. The etiology remains unclear and the diagnosis is based upon patient's clinical picture and anatomopathological findings. Clinically, collagenous sprue is characterized by persistent diarrhoea, severe malabsorption, multiple nutrient deficiencies and progressive weight loss. Differential diagnosis includes celiac disease, which is mandatory to rule out because of their frequent association. Gluten-free diet is the first therapeutic step, but it usually is not effective. However, recent studies show high success rates with immunomodulators, mainly corticosteroids. We report the case of a patient presenting with chronic diarrhea and severe malabsorption who was diagnosed with collagenous sprue, with no response to gluten free diet, but with excellent response to budesonida.
Key words: Collagenous sprue. Chronic diarrhea. Celiac disease. Subepithelial fibrosis. Small bowel.
RESUMEN
El esprúe colágeno es una patología infrecuente del intestino delgado caracterizada por atrofia de la mucosa y depósito excesivo de colágeno a nivel subepitelial. Su etiología es desconocida y su diagnóstico se realiza en base a la presencia tanto de un cuadro clínico compatible como de hallazgos anatomopatógicos sugestivos. Los pacientes suelen presentar diarrea crónica, malabsorción, deficiencias nutricionales graves y una marcada pérdida ponderal. Dentro del diagnóstico diferencial es mandatorio descartar enfermedad celiaca por su frecuente asociación a la misma. La dieta sin gluten es el primer escalón terapéutico, pero generalmente no es efectiva. Sin embargo, estudios recientes señalan altas tasas de éxito mediante el uso de inmunomoduladores. Presentamos el caso de un paciente con diarrea crónica y malabsorción grave diagnosticado de esprúe colágeno, sin respuesta a la dieta sin gluten, en el que el tratamiento con budesonida ha conseguido una excelente respuesta.
Palabras clave: Esprúe colágeno. Diarrea crónica. Enfermedad celiaca. Fibrosis subepitelial. Intestino delgado.
Introduction
Collagenous sprue (CS) is one of the lesser known causes of malabsorption. It is usually associated with persistent diarrhea, weight loss and severe nutritional deficiencies and the diagnosis is clinicopathological (1). CS is a rare but serious disorder, around which there are still many aspects to define, in regard to etiology, diagnosis and treatment. Although celiac disease (CD) is the most common associated problem, CS tends to be increasingly considered as a separate entity (2,3). CS has traditionally been considered as a poor prognosis entity, but there are recent studies that show a good response to treatment with immunomodulators, mainly corticosteroids (3,4). We present the case of a not celiac patient diagnosed with CS with severe clinical involvement who has shown excellent response to treatment with budesonide.
Case report
We present the case of an 84-year-old male with hypertension and chronic hepatitis B under treatment with tenofovir. He referred a 9 month history of watery diarrhea without pathological products, with depositions by day and night and weight loss of 23 kg. He had not had fever, nausea, vomiting, abdominal pain or other symptoms. The physical examination revealed only a malnourished appearance, with body mass index 18.3 kg/m2. Analytically, the only significant changes were hypocholesterolemia (75 mg/dl), hypoproteinemia (4.7 g/dl) with normal proteinogram and INR of 1.3. Renal function, liver function, thyroid hormones, hematological series, vitamin B12, folic acid, ferritin, C reactive protein, immunoglobulins and IgA transglutaminase antibodies were normal. Typing of HLA-II showed absence of alleles associated with CD. Microbiological study of feces (stool culture, Cl. difficile toxin and fresh examination for parasites) was negative. D-xylose test and fecal fat quantification were pathological (2.68 g in urine of 5 hours and 16 g in stool of 24 hours, respectively).
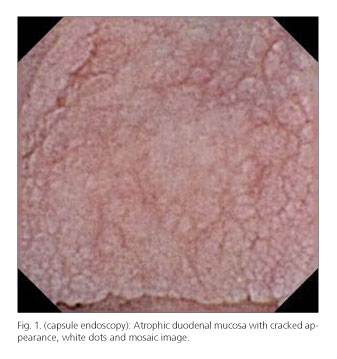
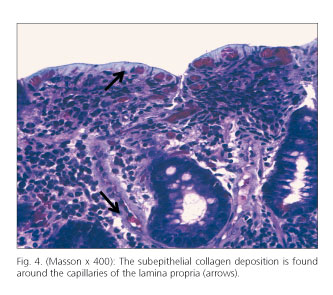
Barium transit was made, appreciating flocculation of contrast in the jejunum. Upper gastrointestinal endoscopy showed an atrophic duodenal mucosa, with cracked appearance (Fig. 1) and complete colonoscopy was normal. Duodenal biopsies showed marked villous atrophy and epithelial denudation (Fig. 2), collagen deposition in a subepithelial layer (Fig. 3) as well as around blood vessels (Fig. 4) and increased lymphoplasmacytic celularity in the lamina propria (Fig. 2), findings consistent with CS (it was not technically possible to determine the thickness of the layer of collagen because electron microscopy was not available). Evaluation of LIE immunophenotype was not suggestive of CD (percentage of intraepithelial lymphocytes (IEL) was not increased, percentage of lymphocytes TCRdδ among total IEL and among total epithelial cells was not increased and percentage of CD3-CD103+ IEL among total IELs was not decreased). Biopsies of the colon, although the association between CS and collagenous colitis has been described, were normal. The study was completed by capsule endoscopy examination, which confirmed marked atrophy of the duodenal and jejunal mucosa, with white dots and mosaic appearance.


CS diagnosis was made and our patient was placed under gluten-free diet (GFD), with improvement of diarrhea and initial gain of 5 kg of weight, but with subsequent relapse with persistent malabsorption, recurrent diarrhea and weight loss of 3 kg again. Thus, despite his advanced age and the presence of chronic hepatitis B virus infection, 3 months after diagnosis he started budesonide 9 mg/d, with good response (disappearance of diarrhea and gain, in a period of 5 months, of 10 kg over the minimum weight registered after recurrence of symptoms) and no evidence of reactivation of hepatitis B (negative viremia before and after initiation of treatment).
Discussion
CS was first described in 1947 and there have been reported about 120 cases since then: It is therefore a very poorly known disorder. It is two-fold more prevalent in women than in men and it tends to occur in middle-aged to elderly patients (1). It is characterized by severe malabsorption with nutritional deficiencies, significant, persistent diarrhea and weight loss that do not usually respond to GFD (3-5). Vomiting and abdominal pain are rare, occurring especially when there is an associated vasculitis. The coexistence of an autoimmune disorder is common and, in fact, it is 2-12 times more frequent than in CD (1).
CS affects the small intestine (mainly duodenum and proximal jejunum) in a patchy way and with variable intensity (1). Severity of symptoms correlates with the overall length of bowel affected rather than with the degree of histological alterations. Intestinal biopsies show subepithelial collagen deposit of variable thickness (both between samples and within the same biopsy) and villous atrophy. The minimum thickness of subepithelial collagen necessary to make a diagnosis of CS has not yet been defined. In a review of 2000 the limit of 12 μm was proposed, establishing also that such deposit should extend into the lamina propria, encompassing cells and blood vessels (6). Another work of 2009 considered that the most appropriate limit was 5 μm (4). Villous atrophy associated with subepithelial fibrosis is usually subtotal or total (Marsh grade IIIb-IIIc) and it is a typical finding the presence of subcryptic inflammation, including formation of crypt abscesses (1,3,4). LIE, unlike CD, are not usually increased, neither show an aberrant immunophenotype nor clonal expansion (3). However, aberrant immunophenotype and clonality of LIE must be ruled out, since CS can also be associated with intestinal lymphoma (6,7). CS diagnosis is clinicopathological: It is based upon the coexistence of a compatible clinical picture and the two typical histological changes (subepithelial collagen deposition and villous atrophy) (1,3). The thickness of the subepithelial collagen layer does not seem to correlate with the clinical severity of CS (3,4).
The etiology of CS remains unknown. The frequent association of CS with autoimmune pathologies and the evidence that it may occasionally respond to corticosteroids (3,4) suggest the immune origin as a probable etiology. Given that this is a malabsorption disorder with villous atrophy, which can show partial response to GFD, and that a large percentage of patients also have diagnostic of CD (8), some authors favor the hypothesis that CS could be a kind of CD difficult to control. Thus, some authors consider CS as a form of refractory CD, which is the one that does not improve clinically or histologically after 6-12 months of strict GFD (after ruling out other causes of villous atrophy). However, there are several data suggesting that CS and CD could be two separate entities. CD serology and the study of HLA alleles associated with CD are negative in a good percentage of patients diagnosed with CS, as it is the case in our patient (3,6). Nevertheless, it should be noted that sensitivity of serology is low in CD adults with villous atrophy corresponding to types I or II of Marsh classification (9) and that serology value can also be limited by a good adhesion to GFD (3). Therefore, in CS patients who present HLA alleles associated with higher risk of CD, the absence of positive CD serology does not rule CS, but rather reflects a not severe degree of villous atrophy or good adherence to GFD. Moreover, although cases of CD without evidence of predisposing HLA are extremely rare, they can constitute a considerable proportion (6 % in European population); so that in the presence of CD positive serology and histological changes compatible with CD, the absence of HLA of risk does not exclude the diagnosis of CD (10). There are also histological differences between CS and CD: CD patients show villous atrophy and hypertrophy of the crypts, while CS is different, with villous atrophy as well as crypt atrophy (6). In addition, intraepithelial lymphocytosis, which is typical of CD, is not usually seen in CS unless it takes place in a CD patient (1). Moreover, it has to be remarked the fact that CS is a disorder that also appears in patients without evidence of CD (3-5), often with an autoimmune problem. This could indicate that CS, instead of a type of CD, could be the intestinal manifestation of a general state of immune dysregulation, which can appear in patients with various autoimmune diseases (2,3). In any case, we must recognize the relationship between CS and CD, since the latter is the autoimmune disease most often associated with CS (3,4).
The frequent presence of subcryptic inflammation suggests an infection as a possible cause of CS. In fact, there have been reports of clinical and histological resolution after antibiotic treatment (11). Finally, CS may be sometimes a paraneoplastic phenomenon, since there are cases of CS in patients with localized colorectal cancer in which tumor resection has also eliminated enteropathy (12).
Some data suggest that CS would involve the entire digestive tract. In some patients, collagen deposition is not limited to small bowel, but also occurs in colon and/or stomach (association with collagenous colitis and/or gastritis) (2-5,13). This deposit appears to come from an increase in collagen synthesis without concomitant increase in its degradation (14).
CS treatment is still controversial. Until recently CS has been conceived as an entity of extremely unfavorable prognosis. GFD usually fails but the possibility of response requires placing it as the first therapeutic step (15). There have not been described predictors of response to GFD but the work of Vakiani et al. found a higher response rate in patients with mild fibrosis compared to those with moderate or severe fibrosis, although the difference was not statistically significant (4). The next step to GFD is the association of immunomodulators, mainly corticosteroids. In recent years very high response rates to these drugs have been described (3,4). Of course, it is essential to provide nutritional support intravenously when necessary.
In the case of our patient, CD was ruled out by the negativity of serology, the absence of HLA alleles associated with CD in the genetic study, the existence of a non-typical histology (crypt hypoplasia, not increased LIE) and the lack of a LIE immunophenotype compatible with CD. As our patient was a man of advanced age, so that he was not the most typical patient with CS, the clinical picture with so marked symptoms and the presence of villous atrophy and subepithelial collagen deposition were the basis for the diagnosis of CS. A history of chronic infection with hepatitis B virus (HBV) could be an interesting factor, given the well known association between such infection and the development of autoimmune diseases like polyarteritis nodosa, autoimmune hepatitis, diabetes mellitus type 1, multiple sclerosis, etc. (16). The possibility that CS could be the intestinal manifestation of a general state of immune dysregulation raises the question of an association or even a possible causal link between the immune alteration secondary to HBV infection and CS. Response to GFD was only transient, so after 3 months we decided to initiate treatment with budesonide, which is a topically acting oral steroid, with few systemic adverse effects. The response has been excellent, with significant weight gain and disappearance of diarrhea.
In conclusion, CS is a rare and severe malabsorptive disorder of unknown etiology, although with a probable autoimmune basis. Its association with CD is undeniable, but nowadays the prevailing opinion is that CS is probably a distinct entity, separate from CD. Diagnosis is clinicopathological and treatment is based on GFD, immunomodulatory drugs (particularly corticosteroids) and sometimes parenteral nutrition. Since the introduction of steroids as an essential part of treatment, prognosis seems much more favorable than in the past. However, many aspects about CS remain still unclear, so that research must continue.
References
1. Zhao X, Johnson RL. Collagenous sprue. A rare, severe small-bowel malabsorptive disorder. Arch Pathol Lab Med 2011;135:803-9. [ Links ]
2. Freeman HJ. Collagenous mucosal inflammatory diseases of the gastrointestinal tract. Gastroenterology 2005;129:338-50. [ Links ]
3. Rubio-Tapia A, Talley NJ, Gurudu SR, Wu TT, Murray JA. Gluten-free diet and steroid treatment are effective therapy for most patients with collagenous sprue. Clin Gastroenterol Hepatol 2010;8:344-9. [ Links ]
4. Vakiani E, Arguelles-Grande C, Mansukhani MM, Lewis SK, Rotterdam H, Green PH, et al. Collagenous sprue is not always associated with dismal outcomes: A clinicopathological study of 19 patients. Mod Pathol 2010;23:12-6. [ Links ]
5. Maguire AA, Greenson JK, Lauwers GY, Ginsburg RE, Williams GT, Brown IS, et al. Collagenous sprue: A clinicopathologic study of 12 cases. Am J Surg Pathol 2009;33:1440-9. [ Links ]
6. Robert ME, Ament ME, Weinstein WM. The histologic spectrum and clinical outcome of refractory and unclassified sprue. Am J Surg Pathol 2000;24:676-87. [ Links ]
7. Medlicott SA, Beck PL, Loken S, Crabtree T. Synchronous collagenous sprue and enteropathy-type T-cell lymphoma: Variants of the same disease. Can J Gastroenterol 2004;18:329-32. [ Links ]
8. Cuoco L. Collagenous sprue with associated features of refractory celiac disease. Rev Esp Enferm Dig 2012;104:223-5. [ Links ]
9. Tursi A, Brandimarte G, Giorgetti GM. Prevalence of antitissue transglutaminase antibodies in different degrees of intestinal damage in celiac disease. J Clin Gastroenterol 2003;36:219-21. [ Links ]
10. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: Results from the European Genetics Cluster on Celiac Disease. Hum Immunol 2003;64:469-77. [ Links ]
11. Stolte M. The histologic spectrum and clinical outcome of refractory and unclassified sprue. Am J Surg Pathol 2001;25:541. [ Links ]
12. Freeman HJ, Berean KW. Resolution of paraneoplastic collagenous enterocolitis after resection of colon cancer. Can J Gastroenterol 2006;20:357-60. [ Links ]
13. Leung ST, Chandan VS, Murray JA, Wu TT. Collagenous gastritis: Histopathologic features and association with other gastrointestinal diseases. Am J Surg Pathol 2009;33:788-98. [ Links ]
14. Daum S, Foss HD, Schuppan D, Riecken EO, Zeitz M, Ullrich R. Synthesis of collagen I in collagenous sprue. Clin Gastroenterol Hepatol 2006;4:1232-6. [ Links ]
15. Holtmann M, von Herbay A, Galle PR, Stremmel W. Langjahrige kollagene Sprue-Remission bei glutenfreier Diat. Z Gastroenterol 1999; 37:1163-8. [ Links ]
16. Maya R, Gershwin ME, Shoenfeld Y. Hepatitis B virus (HBV) and autoimmune disease. Clin Rev Allergy Immunol 2008;34:85-102. [ Links ]
![]() Correspondence:
Correspondence:
Victoria Busto Bea
Department of Gastroenterology
Hospital Universitario Ramón y Cajal
Carretera de Colmenar, Km. 9100
28034 Madrid, Spain
e-mail: victoriabusto@live.com
Received: 19-06-2012
Accepted: 27-07-2012

