










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.105 no.3 Madrid Mar. 2013
https://dx.doi.org/10.4321/S1130-01082013000300010
Esprúe colágeno: no olvidemos el tejido conectivo en la evaluación de la diarrea crónica
Collagenous sprue: Don't forget connective tissue in chronic diarrhea evaluation
Victoria Busto Bea1, Laura Crespo Pérez1, Noemí García Miralles2, Luis Ruiz del Árbol Olmos1 y Ana Cano Ruiz1
Servicios de 1Gastroenterología y 2Anatomía Patológica. Hospital Universitario Ramón y Cajal. Madrid
Dirección para correspondencia
RESUMEN
El esprúe colágeno es una patología infrecuente del intestino delgado caracterizada por atrofia de la mucosa y depósito excesivo de colágeno a nivel subepitelial. Su etiología es desconocida y su diagnóstico se realiza en base a la presencia tanto de un cuadro clínico compatible como de hallazgos anatomopatógicos sugestivos. Los pacientes suelen presentar diarrea crónica, malabsorción, deficiencias nutricionales graves y una marcada pérdida ponderal. Dentro del diagnóstico diferencial es mandatorio descartar enfermedad celiaca por su frecuente asociación a la misma. La dieta sin gluten es el primer escalón terapéutico, pero generalmente no es efectiva. Sin embargo, estudios recientes señalan altas tasas de éxito mediante el uso de inmunomoduladores. Presentamos el caso de un paciente con diarrea crónica y malabsorción grave diagnosticado de esprúe colágeno, sin respuesta a la dieta sin gluten, en el que el tratamiento con budesonida ha conseguido una excelente respuesta.
Palabras clave: Esprúe colágeno. Diarrea crónica. Enfermedad celiaca. Fibrosis subepitelial. Intestino delgado.
ABSTRACT
Collagenous sprue is a rare disease of the small bowel characterized by mucosal atrophy and excessive subepithelial collagen deposition. The etiology remains unclear and the diagnosis is based upon patient's clinical picture and anatomopathological findings. Clinically, collagenous sprue is characterized by persistent diarrhoea, severe malabsorption, multiple nutrient deficiencies and progressive weight loss. Differential diagnosis includes celiac disease, which is mandatory to rule out because of their frequent association. Gluten-free diet is the first therapeutic step, but it usually is not effective. However, recent studies show high success rates with immunomodulators, mainly corticosteroids. We report the case of a patient presenting with chronic diarrhea and severe malabsorption who was diagnosed with collagenous sprue, with no response to gluten free diet, but with excellent response to budesonida.
Key words: Collagenous sprue. Chronic diarrhea. Celiac disease. Subepithelial fibrosis. Small bowel.
Introducción
El esprúe colágeno (ECo) es una de las causas poco conocidas de malabsorción. Suele cursar con diarrea persistente, pérdida de peso y deficiencias nutricionales importantes y su diagnóstico es clinicopatológico (1). Se trata de una patología infrecuente pero grave, en torno a la cual existen todavía muchos aspectos por definir, tanto en lo que se refiere a la etiología como al diagnóstico y al tratamiento. Aunque la enfermedad celiaca (ECe) es el trastorno asociado con más frecuencia al ECo, este tiende a considerarse cada vez más como una entidad independiente (2,3). Tradicionalmente su pronóstico se ha considerado desfavorable, pero existen estudios recientes que muestran una buena respuesta al tratamiento con inmunomoduladores, fundamentalmente corticoides (3,4). En este artículo presentamos el caso de un paciente no celiaco diagnosticado de ECo con afectación clínica severa que ha mostrado excelente respuesta al tratamiento con budesonida.
Caso clínico
Varón de 84 años con hipertensión arterial y hepatitis crónica B en tratamiento con tenofovir. Refería cuadro de 9 meses de evolución de diarrea acuosa, diurna y nocturna, sin productos patológicos, acompañada de pérdida de 23 kg de peso. No había presentado fiebre, náuseas, vómitos, dolor abdominal ni ninguna otra sintomatología. En la exploración física destacaba tan solo un aspecto desnutrido, con índice de masa corporal de 18,3 kg/m2.
Analíticamente las únicas alteraciones relevantes fueron hipocolesterolemia (75 mg/dl), disminución de proteínas totales (4,7 g/dl) con proteinograma normal e INR de 1.3. La función renal, perfil hepático, hormonas tiroideas, series hematológicas, vitamina B12, ácido fólico, ferritina, proteína C reactiva, inmunoglobulinas y anticuerpos antitransglutaminasa IgA fueron normales. El tipado de HLA-II mostró ausencia de alelos asociados a enfermedad celiaca. El estudio microbiológico de heces (coprocultivos, toxina de Cl. difficile y examen en fresco para parásitos) resultó negativo. El test de D-xilosa y la cuantificación de grasa en heces fueron patológicos (2,68 g en orina de 5 h y 16 g en heces de 24 h, respectivamente).
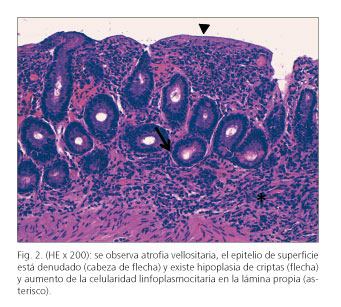
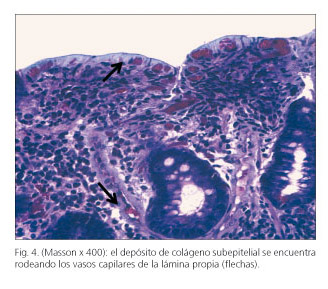
Se efectuó tránsito baritado, apreciándose floculación de contraste a nivel yeyunal. La endoscopia digestiva alta evidenció una mucosa duodenal atrófica de aspecto cuarteado (Fig. 1) y la colonoscopia hasta ciego fue normal. Las biopsias duodenales demostraron marcada atrofia vellositaria y denudación epitelial (Fig. 2), con fibrosis superficial en banda (Fig. 3) y alrededor de los vasos capilares (Fig. 4) y aumento linfoplasmocitario en la lámina propia (Fig. 2), hallazgos compatibles con ECo (no fue técnicamente posible determinar el grosor de la capa de colágeno dado que no se disponía de microscopía electrónica). El estudio inmunofenotípico de las biopsias intestinales no fue sugestivo de ECe (porcentaje de linfocitos intraepiteliales -LIE- no aumentado, porcentaje de linfocitos TCR δ respecto al total de LIE y al total del epitelio no aumentado, porcentaje de linfocitos CD3-CD103+ respecto al total de LIE no disminuido). Las biopsias de colon, si bien se ha descrito la asociación entre el ECo y la colitis colágena, resultaron normales. El estudio se completó con examen mediante cápsula endoscópica, que ratificó marcada atrofia de la mucosa duodenal y yeyunal, con imagen en mosaico y punteado blanquecino.


Se hizo de este modo el diagnóstico de ECo y el paciente comenzó dieta sin gluten (DSG), con mejoría del cuadro diarreico y ganancia inicial de 5 kg de peso, pero con recaí-da posterior con persistencia de malabsorción, recurrencia de la diarrea y nueva pérdida ponderal de 3 kg. Por tanto, a pesar de su avanzada edad y de la presencia de infección crónica por el virus de la hepatitis B, a los 3 meses del diagnóstico se inició budesonida 9 mg/d, con clara respuesta (desaparición de la diarrea y ganancia, en un periodo de 5 meses, de 10 kg sobre el peso mínimo registrado tras la recurrencia de la sintomatología) y sin evidencia de reactivación de la hepatitis B (viremia negativa antes y después de iniciar el tratamiento).
Discusión
El ECo fue descrito por primera vez en 1947 y desde entonces se han comunicado alrededor de 120 casos: se trata, por tanto, de un trastorno muy poco conocido. Es el doble de prevalente en mujeres que en hombres y tiende a presentarse en edades medias-avanzadas (1). Clínicamente se caracteriza por malabsorción grave, con importantes deficiencias nutricionales, diarrea persistente y pérdida ponderal significativa que no suelen responder a DSG (3-5). Los vómitos y el dolor abdominal son raros, presentándose sobre todo cuando aparece vasculitis asociada. La coexistencia de algún trastorno autoinmunitario es común y, de hecho, es hasta 2-12 veces más frecuente que en la ECe (1).
El ECo afecta al intestino delgado (fundamentalmente duodeno y yeyuno proximal) de forma parcheada y con intensidad variable (1). La repercusión clínica, más que con la intensidad de la alteración histológica, se correlaciona con la longitud de intestino afecto. Las biopsias intestinales muestran un depósito subepitelial de colágeno de grosor variable (tanto entre diferentes muestras como dentro de una misma biopsia) y atrofia vellositaria. El grosor mínimo del colágeno subepitelial necesario para el diagnóstico ECo aún no se ha concretado. En una revisión del año 2000 se propuso el límite de 12 µm, estableciéndose, además, que para poder considerar el diagnóstico dicho depósito debía extenderse dentro de la lámina propia englobando sus células y capilares sanguíneos (6). Sin embargo, otro trabajo de 2009 encuentra el límite de 5 µm más apropiado (4). La atrofia vellositaria asociada a este depósito es habitualmente subtotal o total (grados IIIb-IIIc de Marsh) y es típica la presencia de inflamación subcríptica, incluso con formación de abscesos crípticos (1,3,4). Los LIE, a diferencia de la ECe, no suelen estar aumentados; tampoco suele evidenciarse un inmunofenotipo aberrante ni expansión clonal (3). No obstante, estos son siempre aspectos a descartar, dado que el ECo también puede asociarse a linfoma intestinal (6,7). El diagnóstico del ECo es clinicopatológico: se establece ante la coexistencia de sintomatología compatible y las dos alteraciones histológicas típicas (depósito subepitelial de colágeno y atrofia vellositaria) (1,3). El grosor de la capa de colágeno subepitelial no parece correlacionarse con la gravedad clínica del ECo (3,4).
La etiología del ECo continúa siendo desconocida. Su importante asociación con patologías de índole autoinmune y la evidencia de que puede responder a corticoides (3,4) sugieren el origen inmunitario como causa probable. Dado que se trata de un trastorno malabsortivo con atrofia vellositaria, que puede mostrar respuesta parcial a DSG y que un gran porcentaje de los pacientes con ECo tienen además diagnóstico de ECe (8), algunos autores se inclinan por la hipótesis de que se trate de una forma de ECe de difícil control. De este modo, hay quien considera el ECo como una forma de ECe refractaria, que es aquella que no mejora clínica ni histológicamente tras 6-12 meses de DSG estricta (una vez descartadas otras causas de atrofia vellositaria). Sin embargo, hay varios datos que sugieren que podrían ser dos entidades independientes. Así, en un buen porcentaje de los pacientes con ECo la serología de ECe y el estudio de HLA para alelos asociados a ECe son negativos, como es el caso de nuestro paciente (3,6). No obstante, hay que recordar que en celiacos adultos con atrofia vellositaria grado I o II de Marsh la sensibilidad de la serología es baja (9) y además puede verse limitada por una buena adherencia a la DSG (3), de modo que en pacientes afectos de ECo y con HLA asociados a ECe la ausencia de una serología de ECe positiva no permite descartar ECe, sino que más bien refleja un grado no severo de atrofia vellositaria o una buena adherencia a la DSG. Por otro lado, aunque los casos de ECe con ausencia de HLA predisponentes a esta enfermedad son extremadamente infrecuentes, pueden llegar a constituir una proporción no despreciable (6 % del total en población europea), por lo que en presencia de alteraciones histológicas compatibles con ECe y de serología positiva para ECe, la ausencia de HLA de riesgo no excluye el diagnóstico (10). También existen diferencias histológicas entre el ECo y la ECe: los pacientes con ECe muestran atrofia vellositaria e hipertrofia de criptas, mientras que en el ECo existe atrofia tanto de vellosidades como de criptas (6). Además, la linfocitosis intraepitelial típica de la ECe no suele observarse en el ECo a menos que este tenga lugar en un paciente celiaco (1). Por otro lado, y quizá como dato más importante, no puede olvidarse el hecho de que el ECo es un trastorno que aparece también en pacientes no celiacos (3-5), con frecuencia con algún problema autoinmune. Ello podría indicar que el ECo, más que una forma de ECe, podría ser la manifestación intestinal de un estado de disregulación inmune, que aparecería en pacientes con distintas enfermedades autoinmunes (2,3). En cualquier caso, es preciso reconocer la relación existente entre ECo y ECe, dado que esta última es la enfermedad autoinmunitaria que con más frecuencia se asocia al ECo (3,4).
La frecuente presencia de inflamación subcríptica permite pensar en un proceso infeccioso como causa del ECo. De hecho, se han descrito casos de resolución clínica e histológica tras tratamiento antibiótico (11). Por último, también puede tratarse de un fenómeno paraneoplásico, pues existen casos de ECo en pacientes con cáncer colorrectal localizado en los que la resección del tumor ha eliminado también la enteropatía (12).
Algunos datos sugieren que el ECo podría involucrar a todo el tubo digestivo. En algunos pacientes el depósito de colágeno no se limita al intestino delgado, sino que se presenta también en el colon y/o en el estómago (asociación con colitis y/o gastritis colágena) (2-5,13). Ese depósito parece proceder de un aumento en la síntesis del colágeno sin incremento paralelo en su degradación (14).
El tratamiento de los pacientes con ECo es todavía controvertido y hasta hace pocos años se ha concebido como una entidad de pronóstico sumamente desfavorable. La DSG habitualmente fracasa, pero la posibilidad de respuesta exige colocarla como primer peldaño de la escalera terapéutica (15). Aunque no se han descrito factores predictores de respuesta a DSG el trabajo de Vakiani y cols. encontró una mayor frecuencia de respuesta en pacientes con fibrosis leve respecto a los que tenían fibrosis moderada o severa, si bien la diferencia no fue estadísticamente significativa (4). El paso siguiente a la DSG es la asociación de inmunomoduladores, generalmente corticoides. En los últimos años se han descrito porcentajes de respuesta muy altos a estos fármacos (3,4). Por otro lado, siempre que sea necesario habrá que proporcionar soporte nutricional por vía intravenosa.
En el caso de nuestro paciente, la ECe fue descartada por la negatividad de la serología, la ausencia de alelos HLA asociados a ECe en el estudio genético, la existencia de datos histológicos no típicos (ausencia de aumento de LIE, hipoplasia de criptas) y la ausencia de un estudio inmunofenotípico de las biopsias intestinales compatible con ECe. Aunque, dado que era un varón de edad avanzada no se trataba del paciente más típico con ECo, el cuadro clínico con una sintomatología tan marcada y la presencia de atrofia vellositaria y depósito subepitelial de colágeno fueron la base para el diagnóstico de ECo. El antecedente de infección crónica por el virus de la hepatitis B (VHB) podría ser un dato de interés, dada la asociación sobradamente conocida entre dicha infección y el desarrollo de enfermedades autoinmunes como la poliarteritis nodosa, la hepatitis autoinmune, la diabetes mellitus tipo 1, la esclerosis múltiple, etc. (16). Considerando que el ECo podría ser la manifestación intestinal de un estado de disregulación inmune, cabe plantear una asociación e incluso una posible relación causa-efecto entre la alteración inmunitaria secundaria a la infección por el VHB y el Eco. La respuesta a DSG fue solo transitoria; por ello, tras 3 meses se decidió iniciar tratamiento con budesonida por ser un corticoide oral de acción tópica, con escasos efectos adversos sistémicos. La respuesta ha sido excelente, con ganancia ponderal significativa y desaparición de la diarrea.
Como conclusión, el ECo es un trastorno malabsortivo infrecuente y grave, de etiología desconocida, aunque con una más que probable base autoinmune. Su asociación con la ECE es indiscutible; sin embargo, últimamente tiende a considerarse como un proceso independiente. Su diagnóstico es clinicopatológico y su tratamiento se basa en la DSG, los fármacos inmunomoduladores (fundamentalmente corticoides) y, en ocasiones, la nutrición parenteral. El pronóstico, desde la introducción de los corticoides como parte esencial del tratamiento, se perfila de un modo mucho más favorable que en el pasado. No obstante, todavía quedan muchos aspectos por aclarar que hacen necesaria la investigación en este campo.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Victoria Busto Bea
Servicio de Gastroenterología
Hospital Universitario Ramón y Cajal
Carretera de Colmenar, Km. 9100
28034 Madrid
e-mail:
victoriabusto@live.com
Recibido: 19-06-2012
Aceptado: 27-07-2012
Bibliografía
1. Zhao X, Johnson RL. Collagenous sprue. A rare, severe small-bowel malabsorptive disorder. Arch Pathol Lab Med 2011;135:803-9. [ Links ]
2. Freeman HJ. Collagenous mucosal inflammatory diseases of the gastrointestinal tract. Gastroenterology 2005;129:338-50. [ Links ]
3. Rubio-Tapia A, Talley NJ, Gurudu SR, Wu TT, Murray JA. Gluten-free diet and steroid treatment are effective therapy for most patients with collagenous sprue. Clin Gastroenterol Hepatol 2010;8:344-9. [ Links ]
4. Vakiani E, Arguelles-Grande C, Mansukhani MM, Lewis SK, Rotterdam H, Green PH, et al. Collagenous sprue is not always associated with dismal outcomes: A clinicopathological study of 19 patients. Mod Pathol 2010;23:12-6. [ Links ]
5. Maguire AA, Greenson JK, Lauwers GY, Ginsburg RE, Williams GT, Brown IS, et al. Collagenous sprue: A clinicopathologic study of 12 cases. Am J Surg Pathol 2009;33:1440-9. [ Links ]
6. Robert ME, Ament ME, Weinstein WM. The histologic spectrum and clinical outcome of refractory and unclassified sprue. Am J Surg Pathol 2000;24:676-87. [ Links ]
7. Medlicott SA, Beck PL, Loken S, Crabtree T. Synchronous collagenous sprue and enteropathy-type T-cell lymphoma: Variants of the same disease. Can J Gastroenterol 2004;18:329-32. [ Links ]
8. Cuoco L. Collagenous sprue with associated features of refractory celiac disease. Rev Esp Enferm Dig 2012;104:223-5. [ Links ]
9. Tursi A, Brandimarte G, Giorgetti GM. Prevalence of antitissue transglutaminase antibodies in different degrees of intestinal damage in celiac disease. J Clin Gastroenterol 2003;36:219-21. [ Links ]
10. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: Results from the European Genetics Cluster on Celiac Disease. Hum Immunol 2003;64:469-77. [ Links ]
11. Stolte M. The histologic spectrum and clinical outcome of refractory and unclassified sprue. Am J Surg Pathol 2001;25:541. [ Links ]
12. Freeman HJ, Berean KW. Resolution of paraneoplastic collagenous enterocolitis after resection of colon cancer. Can J Gastroenterol 2006;20:357-60. [ Links ]
13. Leung ST, Chandan VS, Murray JA, Wu TT. Collagenous gastritis: Histopathologic features and association with other gastrointestinal diseases. Am J Surg Pathol 2009;33:788-98. [ Links ]
14. Daum S, Foss HD, Schuppan D, Riecken EO, Zeitz M, Ullrich R. Synthesis of collagen I in collagenous sprue. Clin Gastroenterol Hepatol 2006;4:1232-6. [ Links ]
15. Holtmann M, von Herbay A, Galle PR, Stremmel W. Langjahrige kollagene Sprue-Remission bei glutenfreier Diat. Z Gastroenterol 1999; 37:1163-8. [ Links ]
16. Maya R, Gershwin ME, Shoenfeld Y. Hepatitis B virus (HBV) and autoimmune disease. Clin Rev Allergy Immunol 2008;34:85-102. [ Links ]

