










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.105 no.6 Madrid jun. 2013
https://dx.doi.org/10.4321/S1130-01082013000600009
Budd-Chiari syndrome secondary to inflammatory pseudotumor of the liver: Report of a case with a 10-year follow-up
Síndrome de Budd-Chiari secundario a pseudotumor inflamatorio hepático. Seguimiento durante 10 años
Patricia Ramírez-de-la-Piscina, Silvia Estrada, Rosario Calderón, Ileana Duca, Katerina Spicakova, Elvira Delgado, Salvador Zabaleta, Raquel Bengoa and Francisco García-Campos
Department of Gastroenterology. Hospital Txagorritxu. Universidad del País Vasco. Vitoria-Gasteiz, Álava. Spain
ABSTRACT
Inflammatory pseudotumor (IPT) of the liver is a rare benign tumor of unknown origin, it has the appearance of a malignant tumor but has a benign histology and clinical course. We report a case of a 63-year-old man diagnosed of IPT of the liver and followed for 10 years. During the clinical course, he developed a secondary Budd-Chiari syndrome, with a successful response to a transjugular intrahepatic portosystemic shunt over a 5-year follow-up period.
Key words: Budd-Chiari syndrome. Inflammatory pseudotumor. transjugular intrahepatic portal-systemic shunt.
RESUMEN
El pseudotumor inflamatorio (PTI) hepático es una entidad rara, de origen desconocido y aunque puede tener apariencia maligna, su naturaleza es benigna.
Presentamos el caso de un varón de 63 años diagnosticado de PTI hepático en seguimiento durante 10 años. En su evolución desarrolló un síndrome de Budd-Chiari, con buena respuesta tras la colocación de una derivación porto-sistémica transyugular, a los cinco años.
Palabras clave: Síndrome de Budd-Chiari. Pseudotumor inflamatorio hepático. Shunt porto-sistémico transyugular.
Introduction
Inflammatory pseudotumors (IPTs) are rare benign lesions that occurs throughout the body (1,2). Histologically, these lesions are composed of a fibrous stroma and chronic inflammatory infiltrate (1,3,4). Clinically, IPT presents as a progressive space-occupying mass which may be found at different locations. The lung is the most frequently affected organ (3,5). Prognosis is generally good, but despite being a benign lesion histologically, there are some cases of aggressive behaviour and local invasion. Hepatic IPTs are uncommon lesions accompanied by abdominal pain and mass effect. In these cases, a differential diagnosis of hepatocarcinoma or cholangiocarcinoma by radioimaging techniques is an important diagnostic challenge (3,6-8). It was first described, in 1953, by Pack and Baker (2). Since then, more than 200 cases have been reported, in most of them with spontaneous resolution after medical (antibiotics or steroids) or surgical treatment. The association of IPT of the liver and Budd-Chiari syndrome is exceptional and, as far as we are aware, no previous cases have been described in the literature.
The Budd-Chiari syndrome is a clinical condition caused by hepatic venous outflow obstruction (9) and may present as acute, subacute or chronic liver disease. Its etiology is associated with acquired and inherited hypercoagulation states, neoplastic and infectious processes, or space-occupying hepatic lesions. Management of patients with the Budd-Chiari syndrome remains complex despite a variety of treatments for ensuring hepatic venous drainage and preventing postsinusoidal portal hypertension (9-11).
We here report the case of a 63-year-old man diagnosed of hepatic IPT who developed a secondary Budd-Chiari syndrome successfully treated with a transjugular intrahepatic portal-systemic shunt.
Case report
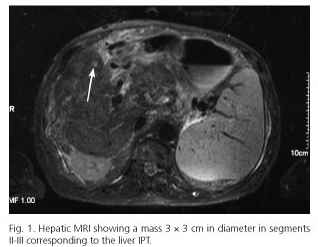
A 63-year-old asymptomatic man was referred to our department in July 2001 because of hepatomegaly (4 cm below the costal margin at the expense of the right lobe) and dissociated cholestasis (serum alkaline phosphatase 873 IU/L and gamma-glutamyl transpeptidase 637 IU/L) found incidentally at routine laboratory tests. Other investigations including antinuclear (ANA), antimitochondrial (AMA), anti-smooth muscle (ASMA) and anti-liver-kidney microsome (LKM) antibodies, porphyria testing, ceruloplasmin, iron profile, alpha-fetoprotein, beta2-microglobulin, carcinoembryonic antigen, hepatitis B virus (HV), hepatitis C virus (HCV) and human immunodeficiency virus (HIV) serological tests, and screening for prothrombotic diathesis were negative or under normal limits. In the study of immunoglobulins, highlight an IgG of 1,900 mg/dl (690-1,400) in connection with monoclonal gammapathy without evidence of myeloma after bone marrow study. An abdominal ultrasound study was unrevealing. The abdominal computed tomography (CT) scan was suggestive of a poorly-defined, hypodense subdiaphragmatic lesion in segments VIII and IV at both sides of the middle suprahepatic vein. The hepatic magnetic resonance imaging (RMI) revealed two lesions, 3 × 3 cm and 3 × 2 cm in diameter in segments IV-VIII and II-III, respectively, on the middle suprahepatic vein and displacement of the right suprahepatic vein (Figs. 1 and 2). Intravascular thrombosis or other lesions in the extrahepatic biliary tree were not observed. It also revealed splenomegaly and left liver lobe hypoplasia. A tentative diagnosis of cholangiocarcinoma and less probable of hepatocarcinoma in a non-cirrhotic liver was established. A fine needle aspiration biopsy was discarded and the patient underwent a diagnostic laparotomy with curative intent. A cystic-appearance mass was found and a core biopsy was obtained. Intraoperative histological examination revealed a fibro-inflammatory process, with presence of lymphoid aggregates, evidence of vascular hyalinization in the absence of microorganisms and atypia, compatible with a PIT of the liver. Because the lesion did not meet criteria of malignancy and considering the high associated morbidity and mortality, resection of the lesion was not performed. The patient was treated with antibiotics and steroids without response.

In January 2006, five years after the diagnosis of hepatic IPT, the patient presented an episode of self-limited upper gastrointestinal bleeding followed by fever, asthenia and anorexia. Physical examination revealed mucocutaneous slight jaundice, bradypsychia, flapping tremor, hepatomegaly and ascites. Laboratory tests showed serum hemoglobin 9.1 g/dL, hematocrit 26.3%, mean corpuscular volume 95, platelets 116,000/mm3, total bilirubin 4.3 mg/dL (conjugated bilirubin 3.8 mg/dL), aspartate aminotransferase 56 IU/L, alanine aminotransferase 106 IU/L, gamma-glutamyl transpeptidase 335 IU/L, alkaline phosphatase 700 IU/L, Gamma 30.7% (10.5-19.5%), leukocytes 7,700 and 800 lymphocytes. Ascitic fluid analysis showed 900 cells/mm3 (polymorphonuclear count > 250 cells/mm3), albumin 0.5, total protein 0.6 g/dL, SAAG > 1.1 and negative culture. The patient was diagnosed of spontaneous bacterial peritonitis and successfully treated with a third-generation cephalosporin. Gastroscopy revealed esophageal varices grade II-III and treatment with beta-blockers was indicated. The abdominal MRI showed chronic liver disease data and how the intrahepatic inferior vena cava was compressed and collapsed by the IPT. A diagnosis of subacute Budd-Chiari syndrome secondary to IPT was established. Treatment with transjugular intrahepatic portosystemic shunt (TIPS) by using self-expanding 10 mm Wallstent was performed, and prompt clinical improvement with resolution of ascites was achieved. The TIPS required no further revisions and was normally patent 5 years after its insertion.
Discussion
The etiology of IPT of the liver remains unclear, although autoimmune disorders (sclerosing cholangitis, autoimmune pancreatitis) or infections (Papillon-Lefevre syndrome, HBV and virus Epstein-Barr infection) have been suggested as causes (12-14). Clinical manifestations may vary from asymptomatic forms to nonspecific symptoms, including fever, abdominal pain or weight loss (4). Our patient presented with an asymptomatic clinical course during the first 5 years of the disease followed by nonspecific symptoms, such as fever, asthenia and anorexia. Conventional radioimaging studies, including abdominal ultrasound examination, CT and RMI are used for the diagnosis of IPT of the liver (5,7,8). In our patient, CT findings were inconclusive but the two liver masses were identified with MRI. Also, because these tumors were not observed in a cirrhotic liver, the presence of hepatocarcinoma was reasonably excluded but given the similarity with other malignant conditions, such as cholangiocarcinoma, and the nonspecific radiological pattern, a diagnostic laparotomy with curative intent was decided (1-6,12,15).
Hepatic IPT are histologically characterized by a fibrous-inflammatory pattern with localized fibrosis proliferation and leukocyte and plasma cells infiltration in the absence of microorganisms proved by tissue culture and atypia. Three histological types of IPT of the liver have been reported: hyaline sclerosis (the pattern described in our patient), plasma cell granuloma and xantogranuloma (2-4).
Most of the cases of IPT reported in the literature showed spontaneous resolution or regression after medical treatment with antibiotic and/or anti-inflammatory drugs. In case of compressive symptoms, surgical excision is the treatment of choice (15). Our patient did not respond to medical treatment with antibiotics or steroids and during the clinical course developed a Budd-Chiari syndrome secondary to compression caused by the IPT. The Budd-Chiari syndrome was successfully controlled with TIPS, and the clinical outcome has been satisfactory during a prolonged follow-up period of 5 years.
By the moment our patient developed a Budd-Chiari syndrome, he shows jaundice, esplenomegaly, thrombocytopenia, caudate lobe hypertrophy and complicated portal hypertension (variceal bleeding, ascites) as signs of biological liver cirrhosis.
It is known that chronically the histological liver damage of Budd-Chiari syndrome may progress to cirrhosis characterized by centrilobular fibrosis and regenerative nodules (16). Liver biopsy was dismissed as clinical, laboratory, radiological and endoscopic data revealed the presence of liver cirrhosis.
In spontaneous bacterial peritonitis, there is a direct correlation between endogenous antimicrobial activity (opsonins activity) and proteins concentration in the ascitic fluid (17). So patients with ascitic fluid proteins below 1 g/dL, as showed our patient, are predisposed to spontaneous bacterial peritonitis (18,19).
In summary, the case here reported illustrates that hepatic IPT is a rare tumor of benign nature with a long-term favourable prognosis but not without potentially serious complications. This is the first report of a patient with IPT of the liver that presented with a secondary Budd-Chiari syndrome, which was successfully treated with TIPS.
Acknoledgements
Thanks to Marta Pulido for her help in translating and editing.
References
1. Fernández de la Puebla Giménez RA, García Alegría J, Lechuga Varona MT, Blanco Molina MA. Seudotumor inflamatorio hepático: a propósito de un caso con respuesta a tratamiento esteroideo. Gastroenterol Hepatol 1999;22:14-7. [ Links ]
2. Pack GT, Baker HW. Total right hepatic lobectomy. Report of a case. Ann Surg 1953;138:253-8. [ Links ]
3. Inaba K, Suzuki S, Yokoi Y, Ota S, Nakamura T, Konno H, et al. Hepatic inflammatory pseudotumor mimicking intrahepatic cholangiocarcinoma: Report of a case. Surg Today 2003;33:714-7. [ Links ]
4. Horiuchi R, Uchida T, Kojima T, Shikata T. Inflammatory pseudotumor of the liver. Clinicopathologic study and review of the literature. Cancer 1990;65:1583-90. [ Links ]
5. Fukuya T, Honda H, Matsumata T, Kawanami T, Shimoda Y, Muranaka T, et al. Diagnosis of inflammatory pseudotumor of the liver: Value of CT. AJR Am J Roentgenol 1994;163:1087-91. [ Links ]
6. Okamoto K, Yoshimi F, Shida D, Oka D, Hagihira T, Shioyama Y, et al. A case of inflammatory pseudotumor of the liver which presented difficulty in differential diagnosis cholangiocellular carcinoma. Jpn J Gastroenterol Surg 2000;33:1900-4. [ Links ]
7. Sakai T, Shiraki K, Yamamoto N, Kawakita T, Ohmori S, Itoh I, et al. Diagnosis of inflammatory pseudotumor of the liver. Int J Mol Med 2002;10:281-5. [ Links ]
8. Flisak ME, Budris DM, Olson MC, Zarling EJ. Inflammatory pseudotumor of the liver: Appearance on MRI. Clin Imaging 1994;18:1-3. [ Links ]
9. Valla DC. The diagnosis and management of the Budd-Chiari syndrome: Consensus and controversies. Hepatology 2003;38:793-807. [ Links ]
10. Nuñez O, de la Cruz G, Molina J, Borrego GM, Marin I, Ponferrada A, et al. Interventional radiology, angioplasty and TIPS in Budd-Chiari syndrome. (Article in Spanish). Gastroenterol Hepatol 2003;28:461-4. [ Links ]
11. García-Pagán JC, Perelló A, Bosch J. Budd-Chiari syndrome. (Article in Spanish). Gastroenterol Hepatol 2000;23:491-7. [ Links ]
12. Akatsu T, Wakabayashi G, Tanimoto A, Kameyama K, Kitajima M. Inflammatory pseudotumor of the liver masquerading as hepatocellular carcinoma after a hepatitis B virus infection: Report of a case. Surg Today 2006;36:1028-31. [ Links ]
13. Kanno A, Satoh K, Kimura K, Masamune A, Asakura T, Unno M, et al. Autoimmune pancreatitis with hepatic inflammatory pseudotumor. Pancreas 2005;31:420-3. [ Links ]
14. Czauderna P, Sznurkowska K, Korzon M, Roszkiewicz A, Stoba C. Association of inflammatory pseudotumor of the liver and Papillon-Lefevre syndrome - A case report. Eur J Pediatr Surg 1999;9:343-6. [ Links ]
15. Seki S, Sakaguchi H, Oiso R, Lee C, Morikawa H, Hamba H, et al. Laparoscopic partial hepatectomy for inflammatory pseudotumor of the liver. Endoscopy 2001;33:294. [ Links ]
16. Ibarrola C, Castellano VM, Colina F. Focal hyperplastic hepatocellular nodules in hepatic venous outflow obstruction: A clinicopathological study of four patients and 24 nodules. Histopathology 2004;44:172-9. [ Links ]
17. Runyon BA, Morrissey B, Hoefs JC. Opsonic activity of human ascitis fluid: A potentially important protective mechanism against spontaneous bacterial peritonitis. Hepatology 1985;5:634-7. [ Links ]
18. Runyon BA. Patients with deficient ascitis fluid opsonic activity are predisposed to spontaneous bacterial peritonitis. Hepatology 1988;8:632-5. [ Links ]
19. Runyon BA. Low protein concentration ascitis fluid is predisposed to spontaneous bacterial peritonitis. Hepatology 1985;5:257-9. [ Links ]
![]() Correspondence:
Correspondence:
Patricia Ramírez de la Piscina.
Department of Gastroenterology.
Hospital Txagorritxu.
C/ José Achótegui, s/n.
01009 Vitoria-Gasteiz, Álava. Spain
e-mail: patrícia.ramirezdelapiscinaurraca@osakidetza.net
Received: 02-10-2012
Accepted: 17-12-2012

