










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.107 n.2 Madrid Feb. 2015
Mutational profile of KIT and PDGFRA genes in gastrointestinal stromal tumors in Peruvian samples
Perfil mutacional de los genes KIT y PDGFRA en los tumores del estoma gastrointestinal en muestras de Perú
José Buleje1, Óscar Acosta1, María Guevara-Fujita1, Yanina Enriquez2, Luis Taxa2,3, Enrique Machicado4, Frank Lizaraso-Caparó1 and Ricardo Fujita1
1 Centro de Genética y Biología Molecular. Facultad de Medicina Humana. Universidad de San Martín de Porres. Lima, Perú
2 Laboratorio de Patología. Facultad de Medicina Humana. Universidad de San Martín de Porres. Lima, Perú
3 Departamento de Patología. Instituto Nacional de Enfermedades Neoplásicas- INEN. Lima, Perú
4 Department of General and Digestive Surgery. Hospital A. Loayza. Lima, Perú
This research has been supported by the Faculty of Medicine of the Universidad de San Martín de Porres (Proyecto N.o E10012008015 "Diagnóstico molecular de cáncer en familias peruanas").
ABSTRACT
Introduction: Gastrointestinal stromal tumors (GISTs) are mesenchymal neoplasms usually caused by somatic mutations in the genes KIT (c-KIT) or PDGFRA. Mutation characterization has become an important exam for GIST patients because it is useful in predicting the response to the inhibitors of receptor tyrosine kinase (RTK).
Objectives: The aim of this study was to determine the frequency of KIT and PDGFRA mutations in 25 GIST samples collected over two years at two national reference hospitals in Peru. There were 21 samples collected from the Instituto Nacional de Enfermedades Neoplásicas (INEN, national cancer center) and 4 samples collected from Hospital A. Loayza.
Methods and materials: In this retrospective study, we performed polymerase chain reaction (PCR) amplification and deoxyribonucleic acid (DNA) sequencing of KIT (exons 9, 11, 13, and 17) and PDGFRA (exons 12 and 18) genes in 20 FFPE (formalin-fixed, paraffin-embedded) and 5 frozen GIST samples.
Results: We report 21 mutations, including deletions, duplications, and missense, no mutations in 2 samples, and 2 samples with no useful DNA for further analysis. Eighty-six percent of these mutations were located in exon 11 of KIT, and 14 % were located in exon 18 of PDGFRA.
Conclusions: Our study identified mutations in 21 out of 25 GIST samples from 2 referential national hospitals in Peru, and the mutation proportion follows a global tendency observed from previous studies (i.e., the majority of samples presented KIT mutations followed by a minor percentage of PDGFRA mutations). This study presents the first mutation data of the KIT and PDGFRA genes from Peruvian individuals with GIST.
Key words: KIT; PDGFRA; Gastrointestinal stromal tumors; Mutational analysis.
Introduction
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors of the gastrointestinal tract (1). The estimated incidence of GIST is 15 cases per million in the general population, and the median age at presentation is 60 years (2). The most common sites of origin for GIST are the stomach (39-70 %) and small intestine (31-45 %), but GISTs may appear anywhere along the gastrointestinal tract or within the abdomen as extra-gastrointestinal tumors (3).
Approximately 95 % of GIST tumors express the tyrosine kinase KIT receptor, also called CD117, which is now accepted as the most specific immunohistochemical marker for GIST (4,5). Other markers, such as CD34 and Desmin that show variable expression (70 % and 5 %, respectively) are also used to confirm the diagnosis of GIST tumors (6-8).
In recent years, it has been established that 75 % to 80 % of GISTs harbor mutations in the KIT gene (c-KIT). Activating mutations of the KIT gene in GIST occur in exons 9, 11, 13, and 17 corresponding to the juxtamembrane (JM) intracellular regulatory domain, the extramembrane domain, and the two intracytoplasmic tyrosine kinase domains, respectively (9). Mutations in the JM domain affect its autoregulatory function and promote spontaneous kinase activation (10). The location of KIT mutations is important for pharmacological treatment; for example, it is well known that a mutation in exon 11 of the KIT gene is associated with a better response to treatment with the inhibitor imatinib (Gleevec®), the potent inhibitor of receptor tyrosine kinase (RTK), and a decreasing response for mutations in exons 9, 13, 17, and wild-type tumors (9).
Another member of the RTK family of genes, PDGFRA, is also associated with the pathogenesis of GIST, and the mutations in KIT are mutually exclusive with those in PDGFRA (11). Mutations in the PDGFRA gene are observed in 7-12 % of cases (12), and the most frequent mutations are observed in exons 12 (regulatory juxtamembrane domain) and 18 (second tyrosine kinase domain).
The presence, nature, and location of the KIT/PDGFRA oncogenic mutations may translate into differences in tumor aggressiveness and influence the likelihood of a clinical response to imatinib, a selective tyrosine kinase inhibitor able to interfere with the activation of KIT and PDGFRA receptors by competing with ATP in the ATP-binding pocket (13,14). Generally, patients with tumors carrying KIT exon 11 mutations respond much better to treatment in comparison with tumors carrying exon 9 mutations (15). However, GISTs with mutations in exon 11 could develop resistance to imatinib treatment if subsequent secondary point mutations appear, which suggests an important escape mechanism for tumor cells with a KIT-dependent proliferation mechanism temporarily inhibited by imatinib (15).
In this retrospective study, we attempted to determine the frequency of KIT and PDGFRA mutations in GIST samples collected from two national referential hospitals in Peru (INEN and A. Loayza Hospital). This is the first study of this type conducted in Peru, and the aim is to encourage a clinical approach on the molecular aspects of GISTs in order to provide adequate treatment for longer survival and a better quality of life for patients with these tumors.
Material and methods
Samples
A total of 25 GIST samples collected over a two-year period were used for this retrospective study. All samples were determined by immunohistochemistry as CD117 positive. Five samples were obtained from frozen tissue, and twenty were derived from formalin-fixed, paraffin-embedded (FFPE) blocks. Coded tumor samples were obtained from two different national reference hospitals (21 from INEN and 4 from A. Loayza Hospital). This study was conducted with an ethical clearance of IRBs of participating hospitals and according to the guidelines of surgical procedures for tumor removal from the Research on Human Subjects Committee policy of the National Health Institute of Peru.
Deoxyribonucleic acid (DNA) extraction
Genomic DNA was extracted using the High Pure PCR template Preparation KIT (ROCHE) following the manufacturer's protocol. A QubitTM fluorometer (InvitrogenTM) was used to quantify the DNA. In 23 samples in which the quality and the quantity of DNA were appropriate, the target exons were amplified. In two of the FFPE tissue samples, the amplification was not successful due to poor DNA quality. The DNA was resuspended in a TE10:1 buffer, coded, and kept at 4 oC until use.
Mutational analysis
Amplification of target exons and mutational analysis of KIT and PDGFRA genes
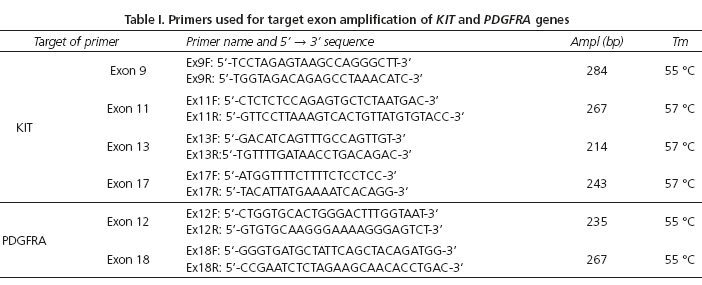
The coding sequence and intron-exon boundaries for exons 9, 11, 13, and 17 of the KIT gene and exons 12 and 18 of the PDGFRA gene were amplified by PCR using primers and the conditions that were optimized in others studies (26,38). The PCRs were performed in a 25 μl total reaction volume consisting of 50 ng of the DNA template. The PCR mix contained 10 pmol of each primer, 0.5 U of Taq polymerase (Thermo Scientific®), buffer 1X, 0.2 mM of each dNTP, and 1.5 mM of MgCl2. The cycling conditions for the PCR amplification were 95 oC for 5 minutes, 35 cycles of 95 oC for 45 seconds, annealing temperatures of 55 oC to 57 oC for 45 seconds (Table I), and 72 oC for 1 minute followed by a final elongation step at 72 oC for 10 minutes. The PCR products were then subjected to electrophoresis on 2 % agarose gels and observed under an ultraviolet light after ethidium bromide staining.
DNA sequencing
PCR amplicons were purified using a QIAquick PCR Purification KIT (QIAGEN) and sequenced in both directions using the BigDye Terminator version 3.1 Cycle sequencing KIT and the 3500 Genetic Analyzer (Applied Biosystems). The generated DNA sequences were analyzed with a Sequencing Analysis software 5.1 (Applied Biosystems) and then aligned using the Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov/Blast.cgi). All mutations were identified based on the National Center for Biotechnology Information database of genetic variation. The reference sequences used to describe the mutations were NM_000222 and NM_0062056 for the KIT and PDGFRA genes, respectively. The numbering of specific mutations and SNPs was referenced from http://www.ensembl.org as of February 2014.
Construction of clones with KIT mutations
For a better determination of the type of mutation (deletion, duplication, and insertion), amplified fragments were cloned. The PCR products were ligated into a PR®8/GW/TOPO® Vector. The construct was transformed into ElectroMaxTM DH10BTM-T1® cells that were grown at 37 oC in an LB medium containing appropriate antibiotics. Plasmid DNA was extracted using the modified alkaline lysis/PEG method and were then digested with EcoR1 to confirm the presence of a fragment. Insert DNAs were sequenced on both strands to confirm sequence variations.
Results
KIT or PDGFRA mutations were found in 21 (91 %) of the 23 samples. In the remaining samples, either no mutation was detected (n = 2), or the sample had a poor DNA quality (n = 2). In total, 18 (86 %) of the 21 samples had a KIT mutation, which were all in exon 11. Only 3 (14 %) samples were mutated in exon 18 of PDGFRA. No mutations were detected in exons 9, 13, or 17 of KIT or in exon 12 of PDGFRA.
The most frequent types of KIT exon 11 mutations were deletions (n = 14), duplications (n = 2), and missense (n = 2) (Table II). Deletions were always in-frame and consisted of 3-27 nucleotides. The most common deletion reported in this work, p.K550_E554del (Fig. 1) was found in 7 tumors (30.4 %), which affected the KIT intron 10-exon 11 splice-acceptor sites and formed a novel intraexonic pre mRNA 3' splice acceptor site. The lack of a wild-type allele (e. g., a mutant allele in homozygosis) was found in 4 samples (17.4 %).
In addition, we identified 4 (17.4%) deletions between codons 557 and 558 (the "hot-spot" region), 2 (8.7 %) deletions involving codon 579, and 1 (4.3 %) deletion between codons 557 and 559. There were two internal tandem duplications involving the 3'-end of exon 11. The lengths of the duplications were 27 pb and 54 bp with codons 571_580 and 574_591 involved, respectively. A missense mutation affecting codon 557 (p.W557R) was detected in 2 samples (Fig. 1). A case-by-case listing of these mutations is shown in figure 2.
We identified 3 samples (14 %) with mutations in the PDGFRA gene involving exon 18, and mutation D842V was detected in 2 (9.5 %) of the 21 samples. In addition, one case (4.7 %) had an in-frame deletion of 4 amino acid' residues at codon 843-846 (12 pb) in exon 18.
In two of the 23 samples, no mutation was detected after analyzing exons 9, 11, 13, and 17 of KIT and exons 12 and 18 of PDGFRA.
Discussion
GISTs are used as a model in the field of molecularly targeted therapies for solid tumors. The almost simultaneous discovery of oncogenic kinase mutations in GISTs and the introduction of kinase inhibitor therapies have led to a rapid evolution in our understanding of these tumors and the biology that defines them (16). The KIT mutation has become more than just a diagnostic marker; it is actually of great value for therapeutic approaches given the development of RTK inhibitors, such as imatinib (17).
In the present study, we have evaluated the spectrum and the frequency of KIT and PDGFRA mutations in 25 GIST samples collected over a two-year period (Table II and Fig. 1). We identified KIT mutations in 86 % and PDGFRA mutations in 14 % of GIST samples, which is similar to previously published ranges (17, 18, 19, and 20). The mutation distribution of KIT exon 11 in the Peruvian sample (86 %) appears to differ from the published data of previous studies ranging between 65.8 % and 72.9 % (1,21). In the case of PDGFRA, this frequency (14 %) was similar to other studies (6 %-35 %) (11,12). The differences in the KIT mutation frequencies regarding other populations can be explained by a possible distinctive genetic profile of the Peruvian samples; however, the reduced sample size may also influence these results.
We have identified seven samples (33 %) with a deletion affecting codons 550-554 (p.K550_E554del), which is a deletion of 15 bp in the end 5' of KIT exon 11 that causes the loss of five amino acids (KPMYE). This mutation affects the juxtamembrane domain of the tyrosine kinase receptor KIT and prevents the constitutive activation of the receptor (30). Another mutation, the deletion p.557_558del, was identified in 4 samples (19 %). The region between the codons 550 and 559 of exon 11 in the gene KIT is the most frequently altered section (17). We found mutations in this region in 89 % of the samples. This region has been reported in previous studies to be associated with the metastatic behavior of GISTs, a poor prognosis (17,23), and a significant reduction of relapse-free survival and represents between 18 %-46 % of all deleted amino acids in KIT exon 11 (24-28). Deletions in the intron10/exon11 junction (codons 550 to 558) have been reported in several studies (16,29) and have been related to a more aggressive behavior of tumors (29). In our study, we have no clinical data to establish that relationship, but it would be a good parameter to measure in future studies (39).
In this study, we did not find mutations in KIT exons 9, 13, and 17, which is consistent with other works (11,31) in which very low mutation rates in these exons have been reported (32,33).
Most of the KIT mutations found in this study were heterozygous (67 %). Lasota et al. (34) showed a strong association between the presence of homozygous KIT exon 11 mutations and malignant clinical outcomes. Tumors with these types of mutations generally had histologic features of sarcomas and developed intra-abdominal and liver metastases in the majority of cases (34). The detection of homozygous KIT exon 11 mutations should be considered to be an additional adverse prognostic marker in GISTs (34).
In the present study, PDGFRA presented mutations in 14 % of the samples (two D842V and one I843_D846del). These samples were exclusively of gastric origin (13 %), which is in agreement with previous reports (7,29). PDGFRA-mutant GISTs have a better prognosis than KIT-mutant tumors. The mutation D842V has been reported to be resistant to treatment with imatinib (22,29) and has been described as the most common alteration in this exon, while the I843_D846del has been reported to respond to treatment with imatinib (35).
In 2006, the U.S. Food and Drug Administration (36) approved a new RTK inhibitor, sunitinib (Sutent®), for the treatment of patients with GIST whose disease has progressed or who are unable to tolerate treatment with imatinib (http://www.fda.gov/bbs/topics/news/2006/NEW01302.html). This recent RTK inhibitor has opened new doors to other RTK-overexpressing tumors in view of the fact that positive therapy responses are obtained in the presence of the wild-type RTK or the previously described imatinib-resistant mutants (37,40).
To date, this is the first report of the molecular characterization of GIST for Peruvian samples. Our contribution will allow patients to have a molecular characterization of GIST that would be useful for a more efficient therapeutic approach in our country.
References
1. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352-9. [ Links ]
2. Judson I, Demetri G. Advances in the treatment of gastrointestinal stromal tumours. Ann Oncol 2007;18(Supl. 10):x20-4. [ Links ]
3. Demetri GD, Benjamin RS, Blanke CD, Blay JY, Casali P, Choi H, et al. NCCN Task Force report: Management of patients with gastrointestinal stromal tumor (GIST) - update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 2007;5(Supl. 2):S1-29. [ Links ]
4. Miettinen M, Lasota J. Gastrointestinal stromal tumors: definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438:1-12. [ Links ]
5. Sarlomo-Rokala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: A sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998;11:728-38. [ Links ]
6. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813-25. [ Links ]
7. Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: A clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005;29:1373-81. [ Links ]
8. Sakurai S, Hishima T, Takazawa Y, Sano T, Nakajima T, Saito K, et al. Gastrointestinal stromal tumors and KIT-positive mesenchymal cells in the omentum. Pathol Int 2001;51:524-31. [ Links ]
9. Tornillo L, Terracciano L. An update on molecular genetics of gastrointestinal stromal tumours. J Clin Pathol 2006;59:557-63. [ Links ]
10. Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008;53:245-66. [ Links ]
11. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708-10. [ Links ]
12. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357-64. [ Links ]
13. Singer S, Rubin BP, Lux ML, Chen CJ, Demetri GD, Fletcher CD, et al. Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol 2002;20:3898-905. [ Links ]
14. Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, et al. STI571 inactivation of the gastrointestinal stromal tumor c-kit oncoprotein: Biological and clinical implications. Oncogene 2001;20:5054-8. [ Links ]
15. Hoeben A, Schöffski P, Debiec-Rychter M. Clinical implications of mutational analysis in gastrointestinal stromal tumours. Br J Cancer 2008;98:684-8. [ Links ]
16. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumors: Origin and molecular oncology. Nat Rev Cancer 2011;11:865-78. [ Links ]
17. Gomes AL, Bardales RH, Milanezi F, Reis RM, Schmitt F. Molecular analysis of c-kit and PDGFRA in GISTs diagnosed by EUS. Am J Clin Pathol 2007;127:89-96. [ Links ]
18. Kitamura Y. Gastrointestinal stromal tumors: Past, present, and future. J Gastroenterol 2008;43:499-508. [ Links ]
19. He HY, Xiang YN, Li Y, Zhong HH, Wu BQ, Zheng J. c-kit and PDGFRA mutations in 60 cases of gastrointestinal stromal tumors (GISTs). Beijing Da Xue Xue Bao 2005;37:320-4. [ Links ]
20. Zheng S, Chen LR, Wang HJ, Chen SZ. Analysis of mutation and expression of c-kit and PDGFRalpha gene in gastrointestinal stromal tumor. Hepatogastroenterology 2007;54:2285-90. [ Links ]
21. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093-103. [ Links ]
22. Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher EC, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumour: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360-7. [ Links ]
23. Wardelmann E, Losen I, Hans V, Neidt I, Speidel N, Bierhoff E, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumours. Int J Cancer 2003;106:887-95. [ Links ]
24. Martín J, Poveda A, Llombart-Bosch A, Ramos R, López-Guerrero JA, García del Muro J, et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: A study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol 2005;23:6190-8. [ Links ]
25. Minárik G, Plank L, Lasabová Z, Szemes T, Burjanivová T, Szépe P, et al. Spectrum of mutations in gastrointestinal stromal tumor patients - a population-based study from Slovakia. APMIS 2013;121:539-48. [ Links ]
26. Tryggvason G, Hilmarsdottir B, Gunnarsson GH, Jónsson JJ, Jónasson JG, Magnússon MK. Tyrosine kinase mutations in gastrointestinal stromal tumors in a nation-wide study in Iceland. APMIS 2010;118:648-56. [ Links ]
27. Steigen SE, Eide TJ, Wasag B, Lasota J, Miettinen M. Mutations in gastrointestinal stromal tumors--a population-based study from Northern Norway. APMIS 2007;115:289-98. [ Links ]
28. Kontogianni-Katsarou K, Dimitriadis E, Lariou C, Kairi-Vassilatou E, Pandis N, Kondi-Paphiti A. KIT exon 11 codon 557/558 deletion/insertion mutations define a subset of gastrointestinal stromal tumors with malignant potential. World J Gastroenterol 2008;14:1891-7. [ Links ]
29. Wozniak A, Rutkowski P, Piskorz A, Ciwoniuk M, Osuch C, Bylina E, et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol 2012;23:353-60. [ Links ]
30. Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 2004;279:31655-63. [ Links ]
31. Lasota J, Wozniak A, Sarlomo-Rokala M, Rys J, Kordek R, Nassar A, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol 2000;157:1091-5. [ Links ]
32. Kinoshita K, Isozaki K, Hirota S, Nishida T, Chen H, Nakahara M, et al. c-kit gene mutation at exon 17 or 13 is very rare in sporadic gastrointestinal stromal tumors. J Gastroenterol Hepatol 2003;18:147-51. [ Links ]
33. Hou YY, Tan YS, Sun MH, Wei YK, Xu JF, Lu SH, et al. C-kit gene mutation in human gastrointestinal stromal tumors. World J Gastroenterol 2004;10:1310-4. [ Links ]
34. Lasota J, Vel Dobosz AJ, Wasag B, Wozniak A, Kraszewska E, Michej W, et al. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab Invest 2007;87:1029-41. [ Links ]
35. Arne G, Nilsson B, Dalmo J, Kristiansson E, Arvidsson Y, Forssell-Aronsson E, et al. Gastrointestinal stromal tumors (GISTs) express somatostatin receptors and bind radiolabeled somatostatin analogs. Acta Oncol 2013;52:783-92. [ Links ]
36. US Food and Drug Administration. FDA approves new treatment for gastrointestinal and kidney cancer (press release). Rockville, MD: FDA; January 26, 2006. Available at: http://www.fda.gov/bbs/topics/news/2006/NEW01302.html. Accessed March 1, 2006. [ Links ]
37. Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, et al. Inhibition of drug resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA 2005;102:11011-6. [ Links ]
38. Went PT, Dirnhofer S, Bundi M, Mirlacher M, Schraml P, Mangialaio S, et al. Prevalence of KIT expression in human tumors. J Clin Oncol 2004;22:4514-22. [ Links ]
39. Garcés-Albir M, Marti R, López-Mozos F, Calabuig-Fariñas S, Navarro S, Ortega J. Results on prognostic value of mutations in localized gastrointestinal stromal tumors (GIST) in one single center. Rev Esp Enferm Dig 2012;104:405-10. [ Links ]
40. Bodega-Quiroga I, Tejedor-Togores P, Sáez-García M, Peraza-Casajús J, Rosado-Dawid N, Serrano-Muñoz A. Tumores del estroma gastrointestinal (GIST): nuevas expectativas de tratamiento. Rev Esp Enferm Dig 2013;105:506-7. [ Links ]
![]() Correspondence:
Correspondence:
José Luis Buleje Sono
Centro de Genética y Biología Molecular
Facultad de Medicina Humana
Universidad de San Martín de Porres
Av. Alameda del Corregidor 1531
Urb Los Sirius, Las Viñas
La Molina, Lima 12. Perú
e-mail: jbulejes@gmail.com
Received: 31-07-2013
Accepted: 10-11-2014

